How Complete Are Your Data?
A look at what the requirements are for "complete data" for laboratories working under the FDA GMP regulations. We also review the observations listed by inspectors on 483 forms from 2006 to 2012, to assess the common mistakes companies make.



United States Food and Drug Administration (FDA) good manufacturing practice (GMP) regulations for quality control laboratories require that laboratory records include complete data derived from all tests. Here, we explore what is meant by "complete data" and, with an analysis of FDA 483 citations, explore the problems that arise when a laboratory fails to understand this phrase. We also consider if complete data include the electronic files generated during the course of the analysis or just the paper printouts.
Reviewing U.S. Food and Drug Administration (FDA) compliance statistics for regulated laboratories is an interesting, albeit somewhat sad, way to spend a Christmas holiday. However, as it's raining stair rods today and will continue to do so for the foreseeable future, the two options available to me are to build an ark or write this column. As I'm not the best handyman in the world, the writing won.
The title of this column is derived from the section of the FDA's good manufacturing practice (GMP) regulations for finished pharmaceutical products specifically concerned with laboratory records — 21 CFR 211.194(a) — which states, "Laboratory records shall include complete data derived from all tests necessary to assure compliance with established specifications and standards, including examinations and assays as follows … ." (1).
Judging from current FDA 483 citations from 2006 to 2012 that are discussed in this column, many regulated quality control laboratories have problems providing complete data during an inspection, which demonstrates that this is a major issue.
Annual FDA 483 Citation Reports
When an FDA inspector pops into a laboratory for tea, biscuits, and a cosy chat, there might be the odd occasion when he or she notices a noncompliance with the way the laboratory operates. (This is never your laboratory, is it?). Every noncompliance that has not been resolved before the conclusion of the audit will be documented on FDA Form 483, which is handed to the site head at the end of the inspection. Copies of these 483s are collated by the agency and published each fiscal year. On the FDA web site there is a page entitled "Inspections, Compliance, Enforcement, and Criminal Investigations" (2). I would like to thank Paul Smith of Agilent Technologies for finding this web page and sharing it with me.
The page is managed by the Office of Regulatory Affairs (ORA) and provides information about the Form 483 observations generated across all of the areas that the FDA regulates, such as drugs, food, and medical devices. If a 483 observation form is prepared by the FDA software application called TurboEIR, any citations in that form are included in the annual spreadsheets available for download at the foot of that web page. There are currently spreadsheets for each FDA fiscal year from 2006 to 2012. However, be prepared:
- The spreadsheets contain citations for all areas that the FDA regulates, so you need to find the tab for the area you are interested in, such as medical devices or pharmaceutical. This way you avoid having to wade through citations dealing with food, although the Capitol Cake Company warning letter (3) can be a diverting and surprising source of entertainment.
- When you open the tab dealing with pharmaceutical companies, the citations need to be sorted to find the area of the Code of Federal Regulations (CFR) you are interested in.
- These spreadsheets are not a comprehensive listing of all inspectional observations because only 483s prepared using TurboEIR are included in the listings; therefore data are not comprehensive but give a representative "snapshot" of compliance, or rather noncompliance, in the pharmaceutical industry.
The area of the CFR that I am interested in is the section covering laboratory records or §211.194. §211.194 is divided into five clauses covering the following topics:
- (a) Testing records
- (b) Test method modification records
- (c) Reagents & standards testing records
- (d) Laboratory equipment calibration records
- (e) Stability testing records
The phrase complete data applies to §211.194(a) and complete records is a consistent requirement for the remaining clauses of §211.194 from subsections b–e.


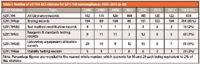
Table I: Number of all FDA 483 citations for §211.194 noncompliances 2006â2012 (4â10)
I have searched the seven fiscal years of 483 observations from the downloaded spreadsheets (4–10) for the number of citations against the clauses in §211.194, and these are presented in Table I. There was a total of 878 citations against §211.194 over the seven years, with the number of citations for noncompliances in any one year ranging from 104 to 145. However, the distribution of the noncompliances per clause was more interesting. The clear area where there were the most problems was §211.194(a): testing records, which constitutes 80% of the noncompliances.
A further analysis of the data within the various subclauses of §211.194(a) shows that three of the major causes of regulatory citations in this area are
- failure to identify the test method used adequately
- failure to record sample weights taken during the analysis
- failure to have the initials of signature of the reviewer.
In essence, these citations are a fundamental failure of either the tester or the reviewer to do their respective jobs correctly. Such failures contrast with the FDA focus on "data integrity" as documented in the scope of the laboratory audit (objective 3) of the "Compliance Policy Guide" 7346.832 on preapproval inspections (11).
In this column, I focus on looking at complete data in connection with testing raw materials, in process samples and finished products. During the course of the discussion, we also look at how the other sections of §211.194 affect complete records for reagents and standards used in testing as well as laboratory equipment and calibration records.
Laboratory Testing Record Requirements
We have seen that the worst area of compliance with the GMP regulations in the regulated laboratory is §211.194(a). This is not surprising because release testing is the major function of regulated quality control (QC) laboratories. However, we need to see what the detailed requirements are for this section. You will recall that earlier I quoted the beginning of §211.194(a) when explaining the title of this "Focus on Quality" column. Following that is a list of eight detailed requirements of the analytical data acquired during the course of analyzing a sample, as shown in Table II, which includes the initials or signature of the person conducting the test. Also required are the initials or signature of a second person checking the analysis to see that data have been acquired correctly, calculations have been performed correctly, and that applicable procedures have been followed. This is the classic "four eyes principle" that we discussed in a recent "Questions of Quality" column (12) for LCGC Europe, a sister publication to Spectroscopy. However, let us remain on track to review the phrase complete data. In Table II, I highlighted the key items for each of the eight requirements of §211.194(a).



Table II: GMP regulations for laboratory data (1)
Please note that there are other sections of §211.194 that also ask for complete data, such as for the standardization of reagents and standards used for tests and for the calibration of our instruments amongst, so you are advised to read §211.194 in its entirety to see the scope of what is meant by complete data. For the purposes of this discussion, we are focusing on the complete data with respect to sample analysis.
Complete Data
You would think that two simple words could not cause a large amount of confusion in regulated laboratories. From a handy dictionary available in my home (13) we can obtain the following definitions:
- complete: having every necessary part or entire
- data: a series of observations, measurements, or facts.
Therefore, my interpretation of complete data from the regulation and the definitions above is the entire series of observations or measurements for a single test. What complete data does not mean is that the analyst makes a selection of the best data that fit his or her testing requirements. This is where subsection 211.194(a) is very explicit, referring to, "A complete record of all data secured in the course of each test, including all graphs, charts, and spectra from laboratory instrumentation, properly identified to show the specific component … , or drug product, and lot tested" (4).
To state it simply, everything that you do — from setting up the spectrometer, to the test itself and any further testing — is covered by the regulation above. You cannot omit anything. You cannot delete anything. So, what does this mean in practice?
The Analytical Process
Let us focus for a few minutes on how we conduct spectroscopic analysis in an analytical laboratory. Samples are taken, stored, and prepared according to the appropriate sampling and analytical procedures and the end result is typically that the samples, standards, blanks, and any quality control standards are ready in vials for analysis. This analytical process is shown in Figure 1. In each stage of the process, records will be generated that are essential for compliance with GMP regulations. Documenting what you have done is a basic tenet of good analytical science. Therefore, documenting laboratory records should be second nature to all analytical scientists regardless of if they work in a regulated laboratory or not.

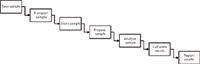
Figure 1: A typical laboratory analytical process.
An alternative process is possible for the identification of raw materials incoming to the warehouse either when a probe is put into the drum or when a handheld device can analyze the contents through the plastic liner of the drum. Typically, the analysis uses a near infrared (NIR) spectrometer located in the warehouse, and the spectrum of the sample is compared with a library that is generated and maintained by the QC laboratory. The data generated in the course of the analysis will be stored and maintained on the instrument's data system along with the laboratory library. Sample information and a record of whether the sample has passed or failed the specification may be generated or entered in a laboratory information management system (LIMS) or enterprise resource planning (ERP) system. However, for the discussion in this column here, let us assume that the analysis is carried out in the laboratory rather than the warehouse.
In the process defined in Figure 1, there will be a sample preparation stage. This step may be as simple as putting a weighed sample aliquot into a volumetric flask and dissolving it, or it may involve a more complex sample preparation scheme. As the samples are prepared for analysis, the analyst will document details about the sample information. This can be done in laboratory notebooks, controlled pro-forma worksheets, or using a LIMS or electronic laboratory notebook (ELN). This information will include batch or lot number, sample identities, sample weights taken, preparation details, dilutions used, and so forth. In addition, the identities of reference standards used and their preparation, the sample analysis sequence and identification of the instrument used in the test also are documented. Although all of this is required by regulation, it is nonetheless good analytical science to ensure that all of these items of data are collected and retained for review (see the items listed in Table II). Documentation of analytical work in this way is an analytical expectation, not an exception, regardless of working in a regulated or unregulated laboratory.
Now we need to prepare the instrument for the analysis, turn the instrument on, and, if appropriate, let it warm up and equilibrate. What happens next depends on how the laboratory has documented its analytical procedures. Typically, there is a system suitability test or point-of-use check performed to verify that the instrument is working within acceptable limits before committing to the analysis of samples. Remember that we are "in the course" of a test, as noted in 211.194(a) (4) in Table II, and that any data present or generated need to be documented and retained. Therefore, even if you are just "evaluating" if the instrument works, the sample or samples used in this process need to be documented and the data generated need to be maintained. Furthermore, the process needs to be consistent with the analytical procedure or standard operating procedure (SOP) that you are following.
Assuming that the reference sample produces acceptable results, the analysis proceeds per the documented analytical procedure:
- Samples are analyzed using the selected instrument, the details of which are recorded in the instrument log book. Any data files generated by the instrument during the course of analysis must be uniquely identified and saved because they are part of the complete data equation as per §211.194(a) (4) above in Table II.
- In addition, the method files, sequence files (if an autosampler is used), processing algorithms, or methods must be documented and also linked to the data files generated.
- The question of paper or electronic records is discussed below in much more detail, but suffice it to say that the electronic records are an essential part of complete data and must be saved regardless of the fact that paper copies of these records are printed out.
- If the analysis is qualitative, then no calculations will be performed on the data and perhaps a statement of identity confirmation is all that is required. However, if quantitative analysis is performed, validated calculations would be used that are typically either within the instrument data system or contained in a separate spreadsheet, a PDF, or additional software. Records of these calculations will be needed as part of complete data.
- Changes to data will be recorded in the audit trail of the data system. These entries are also a portion of complete data.
- Any problems with the analysis may require the samples to be reanalyzed, but only in conformance with the laboratory procedure on out of specification (OOS) results. Again, all sample and standard measurements or spectra are required to comply with the complete data requirement.
- After the analyst has finished his or her portion of the work, it is reviewed and checked by a second individual to check that the work is scientifically sound, the appropriate procedures were followed, data have been calculated properly, electronic records and paper records match, and the audit trail of the data system has been reviewed as another means of ensuring data integrity.
- Finally, when all results are complete and have been checked, a certificate of analysis is generated for the sample and the batch or lot can be released if it is in specification.
This procedure might seem straightforward and simply stating the obvious; however, as we have seen earlier, there are many laboratories that fail — through design or incompetence — to achieve even a basic level of analytical competence as outlined above.
Paper or Electronic Records?
Even before the publication of the FDA's regulation on electronic records and electronic signatures regulations (21 CFR 11) (14), there was a debate about what constitutes the raw data or primary record. Are paper printouts or the underlying electronic records that were used to produce the paper output the primary record or raw data? Now, the FDA has produced a publication that stops the debate once and for all and makes it impossible to justify using paper as the primary record or raw data. Interested? Then read on. I have adapted the rest of this section from a "Questions of Quality" installment in LCGC Europe on defining raw data (15) that some readers may find interesting if they want a wider discussion of this topic.
In 2010, the FDA issued more detailed guidance aimed at chromatography data systems used in QC laboratories that are required to meet GMP regulations. The principles that were outlined, however, can be applied to virtually any computerized laboratory system used for regulated work. This was published on the FDA web site under the concise title: "Questions and Answers on Current Good Manufacturing Practices, Good Guidance Practices, Level 2 Guidance — Records and Reports" (16). Item 3 is a question concerning the interpretation of the GMP predicate rule (1) and the applicability of Part 11 (14) to chromatographic data systems.
The web page posed the question, "How do the Part 11 regulations and predicate rule requirements for GMP apply to the electronic records created by computerized laboratory systems and the associated printed chromatograms that are used in drug manufacturing and testing?" Although the FDA's answer to the question is based on chromatography data systems (CDS), it is applicable to all computerized laboratory systems. If in doubt, take a look at the question that the FDA posed and substitute chromatogram for spectra and the answer is applicable to spectrometers and the associated data systems that generate electronic records. The FDA starts by stating that some in industry misinterpret lines 164 to 171 from the Part 11 Guidance (17) to mean that in all cases paper printouts of electronic records satisfy predicate rule requirements in 21 CFR Part 211. Although aimed at chromatography data, this is applicable to any laboratory data generated especially from spectroscopic systems, therefore in the remainder of this section chromatogram will be changed to spectra.
Therefore, the key to the debate is this: What do the predicate rules state, and how should they be interpreted for a computerized laboratory system? The FDA then comments that for a CDS and other computerized systems used in a QC laboratory involving user inputs, outputs, or audit trials, that there are two clauses from the GMP regulations applicable for the interpretation of paper versus electronic raw data debate. These are §211.68 and §211.180(d) (1).
- 21 CFR 211.180(d) states that manufacturing records must be retained "either as original records or true copies such as photocopies, microfilm, microfiche, or other accurate reproductions of the original records." This clause shows how old the US GMP is because it mentions microfilm and microfiche — the regulation has not been updated since it was issued in 1978 and is firmly grounded in a cellulose world.
- 21 CFR 211.68 further states that "Hard copy or alternative systems, such as duplicates, tapes, or microfilm, designed to assure that backup data are exact and complete and that it is secure from alteration, inadvertent erasures, or loss shall be maintained."
The FDA then makes the following statement, which is reproduced below in its entirety; I have just added the bullet points to aid readability and understanding:
- The printed paper copy of the spectra would not be considered a "true copy" of the entire electronic raw data used to create that spectra, as required by 21 CFR 211.180(d).
- The printed spectra would also not be considered an "exact and complete" copy of the electronic raw data used to create the spectra, as required by 21 CFR 211.68.
- The spectra does not generally include, for example, the injection sequence, instrument method, integration method, or the audit trail, of which all were used to create the spectra or are associated with its validity.
- Therefore, the printed spectra used in drug manufacturing and testing do not satisfy the predicate rule requirements in 21 CFR Part 211. The electronic records created by the computerized laboratory systems must be maintained under these requirements.
Why the lengthy debate on this point? The reason is that electronic records contain much more information than the corresponding paper printouts of the same spectroscopic run. Consider an Excel file as a simple example, under the "properties" tab there is information about the file creation and printing dates that a typical printout does not contain unless specifically configured to do so. In this simple case, the electronic record contains more information than the paper one. Now think about an analytical run. Who prints out the audit trail? No one. However, the audit trail contains critical information to demonstrate the integrity of the analytical run and the conversion of the raw data files into the final reportable value and also identifies critical operator interactions that are interpreted and used to modify the data and carry out calculations. Therefore, the electronic records from a data system contain much more that just the paper printout. That is why the FDA wants electronic records. Printed spectra do not satisfy the predicate rule requirements in 21 CFR 211.
In summary, a paper printout does not meet the requirements of the predicate rule and a spectrometer system must therefore be considered either a hybrid system (electronic records with signed paper printouts) or a fully electronic system. Regardless of the way a spectrometer system is used (hybrid or homogeneous system), the electronic records must be maintained and are key to meeting the predicate rule requirements.
However, I think the FDA missed a trick in this discussion because the key regulatory requirement for the QC laboratory is §211.194(a) (1) (Table I), which states that, "Laboratory records shall include complete data derived from all tests necessary to assure compliance with established specifications and standards." For example, an audit trail is part of the complete data regardless of whether the system is a hybrid or fully electronic one. Therefore, if you use the argument that your laboratory records are paper, then you do not have a leg to stand on, because paper records can never be complete.
As a result of several cases of fraud and falsification identified over the past few years by the agency, all FDA inspectors have undergone training in data integrity by Monica Cahilly (Green Mountain Quality Assurance). Now, during inspections they will focus on the system and the electronic records contained within it rather than the paper outputs. Paper is now incidental to the integrity of the underlying electronic records generated by any computerized laboratory system used in a regulated laboratory.
Note that complete data also include the audit trail entries created during the generation, interpretation, and modifications that occur during an analysis.
Generating PDF Reports Instead of Paper?
One question arising from the discussion of paper versus electronic records is the use of PDF output as a substitute for the printout, here the PDF is just used as a report not as an archive medium. Let's discuss this further. If there is only a PDF report and no information about the sample information, sample weights, details of the preparation of reagents or standards, and no spectroscopic data files, the report is wrong and incomplete. This is exactly the same argument that the FDA shot down in flames for a paper report, as discussed in the last section. The only difference is that instead of paper output the same information is on an electronic report.
The only time, in my view, that a PDF report is acceptable as part of the complete data for an analysis is when
- The PDF report is generated securely, following a documented, secure, and controlled process (this is required so that the information used to generate the PDF cannot be selectively edited or "enhanced" during the process). Visibility and traceability are key, because the problem with PDF files is that it is possible to open unprotected and even some protected PDF files in the professional version of Acrobat, make changes to the content and then save them. PDF document formats are integrated into Microsoft Office applications that can be used to open, extract the data, edit the files, and then re-render them into a PDF. Even photocopiers include the ability to copy a spectra and generate a PDF. So, in an insecure process, the information on a printed paper spectra (such as peak area) could be edited and the data copied to PDF.
- After they are generated, PDF reports must be protected throughout the retention period. This can either be within the laboratory application that generated the file, in a document management system, or on a protected directory on a network. The aim is to avoid losing the file from a disk crash or indeed edit the file subsequently.
- The underlying laboratory records used to generate the report must be linked to the report, for example, data files from which the result information has been abstracted, audit trail entries, methods and calculations. Even if "final" records are electronically embedded within the report (such as integrated spectra), the PDF needs to provide traceability to the electronic source file. Furthermore, it is essential that the raw data files or electronic records be available in case reinterpretation is required in the case of a laboratory investigation or a regulatory challenge.
Therefore, a laboratory relying on a PDF report file is totally inadequate because it does not comply with the regulations for complete data, even if printed or PDF copies of the underlying spectra are attached. Think this through with all of your laboratory operations: What happens if you use a third-party supplier to do your instrument qualification? What output from the process could you get? A PDF report (either on paper or electric version) and the service engineer walking out of the laboratory with the data files on his or her PC? Try explaining this to an inspector. How much of an idiot do you wish to appear to be?
As well as the need to retain the electronic data, the data flow used by the engineer from the start of the process to the generation of the PDF report (and the traceability of any imbedded electronic information) must be clearly understood by you — because you will need to defend it during an audit. Do you ever look at what the engineer has done? Have you looked at the raw data to see how many attempts have been made to get the instrument to pass? No? I thought not.
Europe Has New GMP Regulations for QC Laboratories!
Although this column has focused exclusively on the FDA GMP regulations for regulated laboratories, we also need to consider that we work in a global environment and there are other regulations to consider. At the start of July 2013 the new version of European Union GMP Chapter 6 for Quality Control (18) will go into effect. Here, the main changes are the inclusion of a new section on technical transfer of testing methods and other items such as out of specification results. (There are five new clauses specifically on method transfer.)
Although essentially similar, there are some differences between the US and EU regulations for QC laboratories such as:
- 6.9: Some kinds of data (such as tests results, yields, and environmental controls) should be recorded in a manner permitting trend evaluation. Any out of trend or out of specification data should be addressed and subject to investigation. Therefore, data must be trended under EU GMP, but this is only contained in the "OOS Guidance for Industry" from the FDA (19).
- 6.10: In addition to the information that is part of the batch documentation, other raw data such as laboratory notebooks or records should be retained and readily available. The EU regulation is implicit in requiring complete data and is not as detailed as the FDA GMP.
Although the section on laboratory records in Chapter 6 is relatively brief compared with the 21 CFR 211, there are some further requirements of the batch records required in Chapter 4 on documentation (20).
Summary
In this column we have looked at what the requirements are for "complete data" for laboratories working under the FDA GMP regulations and also looked at the mistakes that are made by reviewing the 483 observations made by inspectors over 2006–2012. The problem is that elementary analytical science mistakes are being made by staff and laboratory management that are due to poor education, laziness, or incompetence. Judging from the FDA's data nothing appears to be getting better and are becoming worse as the inspectors are looking harder and in more detail especially after their data integrity training. So in the short term, things will get worse in terms of the number of citations that will rise before regulated laboratories get the message and improve. We have also looked at whether paper printouts or electronic records are adequate to meet GMP regulations and, surprise, it is not paper.
References
(1) Current Good Manufacturing Practice for finished pharmaceutical products, 21 CFR 211, 2009
(2) US Food and Drug Administration web site, "Inspections, Compliance, Enforcement, and Criminal Investigations," page http://www.fda.gov/ICECI/EnforcementActions/ucm250720.htm.
(3) FDA Warning Letter, Capitol Cake Company, August 2008.
(4) FY 2006 Inspectional Observation Summaries – download from reference 2.
(5) FY 2007 Inspectional Observation Summaries – download from reference 2.
(6) FY 2008 Inspectional Observation Summaries – download from reference 2.
(7) FY 2009 Inspectional Observation Summaries – download from reference 2.
(8) FY 2010 Inspectional Observation Summaries – download from reference 2.
(9) FY 2011 Inspectional Observation Summaries – download from reference 2.
(10) FY 2012 Inspectional Observation Summaries – download from reference 2.
(11) FDA Compliance Program Guide 7346.832 Pre-Approval Inspections (published in 2010 but effective from 2012).
(12) R.D. McDowall, LCGC Europe 24(4), 208–216 (2011).
(13) Collins Dictionary & Thesaurus (William Collins, London, 1988).
(14) Electronic Records; Electronic Signatures final rule (21 CFR 11) (1997).
(15) R.D. McDowall, LCGC Europe 25(4), 194–200 (2011).
(16) Questions and Answers on Current Good Manufacturing Practices, Good Guidance Practices, Level 2 Guidance — Records and Reports. Part 3 (2010) http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm124787.htm.
(17) FDA Guidance for Industry, Part 11 Scope and Application, 2003.
(18) EU GMP Chapter 6 (2013).
(19) FDA Guidance for Industry, Out of Specification Results (2006).
(20) EU GMP Chapter 4, Documentation.
R.D. McDowall is the principal of McDowall Consulting and the director of R.D. McDowall Limited, and the editor of the "Questions of Quality" column for LCGC Europe, Spectroscopy's sister magazine. Direct correspondence to: spectroscopyedit@advanstar.com



R.D. McDowall















