Periodic Reviews of Computerized Systems, Part II
Continued discussion of the periodic review process, including how to conduct a review, the use of checklists, and reporting the outcome



This column, which is the second of a two-part series, looks in more detail at a periodic review.Here, we will discuss how to conduct the review, the use of checklists, and reporting the outcome.
In part I of this column (1), we discussed what a periodic review is, how to define the scope of a system scheduled for a review, and the main elements in the planning and execution of a review. In the second part of this discussion on periodic reviews, I want to focus on the execution phase of a periodic review and discuss in more detail the following activities:
- the opening meeting
- carrying out the periodic review
- reviewer's closed meeting
- the closing meeting
- writing and approving the periodic review report and action plan
- implementing corrective and preventative actions.
Who Is Involved and What Do They do?
Who should be involved with a periodic review, apart of course, from the person who is conducting the review (as we discussed in part I of this column)? Ok, here is a list of potential people. Some of these roles may be combined depending on the size of the system being reviewed and especially if the laboratory is responsible for the whole system including the IT support, if the system is relatively small, or if there are only a few users. Other persons may be involved, depending on the roles and responsibilities for the system:
- Laboratory manager: Although the laboratory manager may not be directly responsible for the system itself, the system operates in the laboratory for which the manager is responsible and accountable. At a minimum, the laboratory manager should be present at the opening and closing meetings to introduce him or herself and listen to the review conclusions and discuss any findings.
- Process owner: The individual in the laboratory who is responsible for the system. For a standalone spectrometry system, this may be the spectroscopist who runs the system. For larger systems, say the high performance liquid chromatography–mass spectrometry (HPLC–MS) networked system that was discussed in the first part of the column (1), it may be that the process owner is the laboratory manager.
- System owner: The system owner is the person responsible for supporting the system from an IT perspective. If the system is standalone (where IT support comes from within the laboratory), the role of system owner can be taken by the process owner. If the the system is networked, the system owner may be an individual in the IT department. In the latter situation, the system owner could be from an external company if IT operations have been outsourced.
- Power user or laboratory system administrator and users: Depending on the size of the system, these roles could be taken by a single person or by many individuals. However, the reviewer needs to have access to somebody within the laboratory who knows the system in-depth from a technical perspective (the power user or system administrator) and may also require an experienced user to discuss procedures and use of the system.
- Computer validation: Because the periodic review will examine the last validation of the system, there needs to be a person available to answer the reviewer's questions on this topic. Depending on how the work was performed, this could be the system administrator from the laboratory or a member of the computer validation group within the organization.
- Quality assurance (QA): A member of QA who is responsible for the laboratory may also be involved with the periodic review. Depending on the depth of this person's knowledge, his or her involvement may range from interested observer to full participant.
Here is where the timetable for the periodic review comes in. Instead of having everybody hanging around waiting to be called, a timetable for the review should state what topics will be covered and roughly when they will take place. This allows individuals to carry out their normal jobs and turn up at an appointed hour. There may be some variation in the timetable depending on what the reviewer finds, so prepare to be flexible with the timing.
Types of Computerized Systems to Be Reviewed
Typically, there are two types of computerized systems that will be covered by a periodic review: hybrid systems (electronic records plus paper printouts with handwritten signatures) and homogeneous systems (electronic records with electronic signatures where the only printouts will be summary sheets or at the request of an inspector or reviewer). Let's look at the advantages and disadvantages of each, starting with a completely paper-based system as shown in Table I. Paper is a medium that we are all familiar with; it is tangible, we can handle it and pick it up. Also, the audit trail is apparent because the paper is written or printed in a linear fashion. However, the poor reviewer has a disadvantage as he or she must wade through piles of paper to understand what was done, by whom, and when.


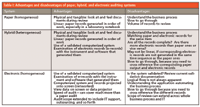
Table I: Advantages and disadvantages of paper, hybrid, and electronic auditing systems
Moving to a hybrid computerized system, we have the same advantages and disadvantages of the paper plus the complication and correlation of electronic records. It is the correlation of the paper with the electronic records that provides the biggest challenge in an inspection or periodic review of heterogeneous or hybrid systems. The failure by Food and Drug Adminstration (FDA) inspectors to cross check the paper printouts with the corresponding electronic records in a chromatography data system allowed Able Laboratories (Cranbury, New Jersey) to conduct fraud on a large scale for a long period of time (2). If an electronic (homogeneous) system is audited, the reviewer needs to ensure first that the computerized system itself is validated and has remained so from the last full validation to the time of the periodic review. However, the reviewer needs an experienced user when checking the way the system operates and how the electronic records are produced to implement the requests from the reviewer through the system. Often, the system used for part of the review may use the validation or training instance and the reviewer needs to ensure that the operational and other instances are equivalent. However, the great advantage when reviewing an electronic system is the speed of the audit; a lot of ground can be covered relatively quickly because of how the system supports the business process. What also needs to be addressed are the technical controls within the system to ensure data integrity, for example, checking the audit trail to make sure that results have not been manipulated to pass. As an electronic system can cross organizational departments, the review scope may be wider than normal and if networked will also include the IT department responsible for supporting the system.
Are Computerized Systems Designed to Help Periodic Reviews?
No, is the simple answer to a simple question. Let us explore the reason why we want systems to help us with periodic reviews.
(Turn on moan-mode). Much of the emphasis in the design of the majority of computerized systems used in the analytical laboratory has focused, unsurprisingly, on providing new functions for users and incorporating regulatory compliance, such as audit trails when requested by regulated users. However, as we are moving toward electronic systems with minimal paper output, we really do not have the functions in software to allow effective and efficient second-person review of the data on a day-to-day basis and also for periodic reviews on a 1–3 year frequency. It is the need for second-person review that we need to concentrate on because the periodic review will use the same functions. Both the second-person check and the periodic reviewer need to assure themselves that the data quality and integrity are good and have not been compromised. This is also a concern for regulatory agencies, especially the FDA, which recently launched an initiative on ensuring data integrity (3).
The United States good manufacturing practices (US GMP) regulations require, in section § 211.194(a), that laboratory records include complete data derived from all tests necessary to assure compliance with established specifications and standards, including examinations and assays, as follows:
(4) A complete record of all data secured in the course of each test, including all graphs, charts, and spectra from laboratory instrumentation, properly identified to show the specific … drug product, and lot tested.
(8) The initials or signature of a second person showing that the original records have been reviewed for accuracy, completeness, and compliance with established standards (4).
These two clauses are important because the regulation requires that all data be collected. This includes the checks carried out on the spectrometer before sample analysis commences, as well as any rerun samples. The checks performed by the reviewer (second person) must include complete data, including audit trails of hybrid and electronic computerized systems.
Referring to Table I, the complete data should be present in the laboratory notebook or uniquely numbered laboratory sheets and instrument printouts. Built into the paper will be the cellulose audit trail — that is, where mistakes that have been identified and corrected will have the original entry struck through with replacement data entered by the side, the initials of analyst who made the change, and the date of the change. The correction may also include a reason for change as per the applicable good X practice (GxP) regulation that the laboratory works under.
When we consider hybrid systems, our data integrity problems start because the audit trail becomes jumbled and the linkage between electronic records and the corresponding paper printouts is not always obvious. Often, the audit trail is not very good as there is just a single audit log with little or no search function. By its nature, an audit trail will normally contain a multitude of entries: user access, failed attempts for access by users, additions, modifications, and deletions to data, user account management and access control, and so forth. Many of these entries are routine events, but unless there is a simple and easy means of separating different types, the audit trail can be useless.
Similarly, when fully computerized systems are used, the linkage between records and reports is usually easier and more straightforward. However, the key requirement is an effective audit trail, which may not be the case. Typically, systems are not designed to work electronically: The business process may be fine but the Achilles heel of the majority of systems is the audit trail as it is the key to ensuring data integrity. What is required are data filters that can be configured by users to identify when changes to data have been entered into the audit trail that will allow effective second-person review and, by implication, periodic review. In addition, there is the new European Union (EU) GMP requirement (5) for review of audit trails and few if any systems have the ability to demonstrate that this has been done. So, we are left with systems that are typically not designed to facilitate reviews of data, audits, or inspections. Welcome to the periodic review world of hybrid and electronic computerized systems! (Turn moan-mode off.)
Opening Meeting
So having moaned about the systems you have to review, let's begin the process. This starts with the opening meeting, which is typically a short session where the reviewer meets the key people who will be involved with the review. It allows introductions to be made and for the reviewer to explain what the aims of the periodic review are and set expectations from the individuals who will be contributing from the laboratory side. It also provides an opportunity to fine-tune the schedule to take into account any last-minute availability of staff and to move items around. The point here is that the reviewer should be flexible and, so long as the main topics are covered, the order of review should not matter.
One item that is important is at the end of the opening meeting and before the start of the review, the reviewer should ask for a short presentation of the system by the process owner or validation manager to outline the main functions and how the system was validated. This allows the reviewer to understand the context of the material he or she has read before the review starts and also to pick up and understand terminology.
With Cat-Like Tread?
I want to consider how you actually carry out a periodic review. In my view, you have two basic options:
- Static: Sit in a room and read and evaluate system documentation or logs against procedures and polices. At this point, the more intelligent readers among you may be wondering if watching paint dry is a more intellectually stimulating option.
- Dynamic: Look at the system and see how the software and instrument operates, examine the IT support, and have discussions with users and IT staff. You will be amazed at what you can find out by simply being observant, which we will discuss later in this column.
In reality, the periodic review is a mixture of the dynamic and static options. Walking around the laboratory and seeing the system in its operational environment allows a reviewer to put the documentation into context and see the reality rather than what is portrayed in policies and procedures. So, the reviewer should not just sit in an office and read — walk around and see the system in operation in the laboratory. Therefore, the review schedule should contain time for a tour of the laboratory and system and, if applicable, the IT department.
Conducting the Periodic Review
You will need to obtain information from the people that you will interview and have discussions with during the audit. The best way to do this is to ask open-ended questions (for example, when? who? where? how? why? and what?) to get people talking. Then, of course, comes the question that makes you ask if all periodic reviewers are from the state of Missouri. Because the answer to the open questions listed above is usually followed by the infamous "show me" to verify that what has been stated actually occurs and there is documented evidence available to demonstrate this. To verify the information provided, the reviewer should use closed questions that the responder can respond to with either yes or no answers. Another technique that allows a reviewer to understand information is to ask questions such as, "What happens if … ?"
Some of the dos and don'ts of conducting periodic reviews are shown in Table II, and many of these are derived from how to manage people correctly during the review. Part of the role of a reviewer could be to persuade users and IT staff to change their way of working if noncompliant practices are observed; you can't do this if you have put their backs up when you conduct the review.



Table II: The dos and don’ts of conducting a periodic review
I've Got a Little List
If you want to strike terror into the individuals involved in a periodic review, a very good way at the opening meeting is for the reviewer to casually open a briefcase and pull out the most devious instrument of torture imaginable. Like a magician pulling a rabbit from a hat — out comes … the checklist! Moreover, the thicker the checklist and the louder the sound it makes when it hits the table, the greater the terror perceived by the victims.
Seriously though, a checklist has a number of advantages for a reviewer. It should have been generated from the applicable regulations, pharmacopeial general chapters, company procedures, and regulatory and industry guidance documents. However, as the regulations are changing at a rapid rate, the checklist should be a living document and be versioned and dated to ensure that the current version is being used by the reviewer. As such, the aim of the checklist is to ensure consistency across the company, as well as between sites and reviewers, and that all aspects of the review have been covered. From the perspective of the reviewer, a checklist is a memory aid to help them cover all the points.
Death by Checklist?
There is also a downside to a checklist: A reviewer can be a slave to it. Imagine, if you can, the scene during a periodic review where the reviewer asks the following: "Question 107: Have you got a backup SOP? Question 108: Have you kept a log of backups performed? Question 109: …" and so forth. At this point, the poor individual on the receiving end of this inquisition is mulling over the philosophical question: Which is better, murder or suicide?
This situation can be real if a reviewer takes the checklist literally. Instead, the checklist should be used as a guide and, if structured correctly, will be broken up into different subject areas that, when taken together, will cover all applicable topics. When a specific topic is covered, the reviewer should ask questions and discuss with the individuals involved. The reviewer just needs to refer to the checklist to ensure that everything has been covered before moving on to another topic.
Furthermore, as mentioned in part I of this series, the reviewer needs to be aware that if something important is found during the review, it may warrant a more detailed investigation. In such a case, a portion of the review may be expanded and other parts not covered.
Options for Checklists: Working Smarter Not Harder
Let me ask you a personal question: Are you lazy? We have looked at checklists, but we also need to consider how to use them effectively throughout the periodic review process. Rather than just having a list of questions that are ticked off as the review progresses, we need to think how we can link the checklist with the review report. Indeed, can a checklist form the basis for the audit report? The answer is yes. One way of doing this is shown in Table III. Here, in the left-hand column are the checklist questions and in the right-hand column is space to write in the observations and any findings or recommendations (we will return and define these terms later in this column). However, you can see that with a little planning, the checklist and report can be integrated easily and the checklist becomes the basis for the periodic review report.



Table III: Advantages and disadvantages of auditing paper, hybrid, and electronic systems
Review of the Last System Validation
It is ironic that computer systems that are supposed to automate operations and streamline processes generate mountains of paper. So if a system subject to a periodic review has been validated since the last review, the current review should start with the last system validation. Depending on the size and complexity of the computerized system, this could take a short time or last at least a day. So where do we begin?
Rather than list my ideas, let's hear from the GMP inspectors who do this for a living. Sections 23 and 24 of PIC/S guide PI-011, called computerized systems in GXP environments (6), are devoted to the inspection of computerized systems in regulated environments. This guide has also been adopted by the good clinical practice (GCP) inspectors in the EU (7), rather than write their own. This is great, except that the PIC/S guide is outdated, being based on good automated manufacturing practice (GAMP) 4 (8) instead of GAMP 5 (9), leading to very little risk-based computerized system validation and little flexibility in the approach.
However, PIC/S PI-011 section 24 (6), entitled "Inspector's Aide Memoires," contains six checklists for inspecting a computerized system although the main focus of these checklists is on the application, there is little on the IT infrastructure and services supporting it. Section 23, entitled "Inspection Considerations," is more interesting from the perspective of a periodic review and how to conduct one, although clause 23.4 notes that this guidance, much the same as any checklist, should not be used as a blunt instrument when inspecting a computerized system, but used selectively to build up an understanding of how computerized systems are validated and operated by a company. However, we can learn and adapt the approach outlined here.
Preparation Before the Review
As mentioned in the first part, the reviewer needs to do a little homework by reading some key documents before he or she starts the periodic review. The PIC/S guide suggests starting with the validation summary report (6); however, in my view this is only adequate, and a better approach is to read the following documents:
- Validation plan — the last full validation plan of the system will detail the activities and documented evidence that were intended to be performed and written, respectively.
- Validation summary report — to see what was actually done and understand the reasons for the differences and the justification for the changes made to the plan.
- User requirements specification (URS) — to define the intended use of the system and to assess the quality of the requirements: Are they testable or verifiable?
The rationale for reading these documents is shown in Figure 1. It checks the quality of the validation control by comparing both the intent and delivery of the validation effort and asks if they match; this provides a good foundation to begin the review of the validation of the system. If the validation is suspect, then the quality and accuracy of the results produced by the system will be suspect as well. Reading the URS determines how well the intended purpose of the system has been defined and, hence, how well the rest of the validation, shown with dotted boxes in Figure 1, is likely to have been performed.


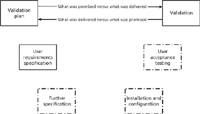
Figure 1: Preparatory reading before a periodic review.
Reviewing Requirements: Role of Traceability
You may remember that I discussed the role of a traceability matrix in validation in a two-part column a few years ago (10,11). These articles will provide you with the background to what I'm going to discuss now from the perspective of a periodic review. The traceability matrix should allow a reviewer to trace requirements from the URS to where the requirement is either verified or tested later in the life cycle or vice versa (test or verification back to the requirements). So, what if there is no traceability matrix? Well in essence, I ask the questions and then watch you wade through a sea of paper to find the answer. This is a form of entertainment for the reviewer but only if he or she can keep from smiling.


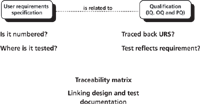
Figure 2: The importance of being able to trace requirements to testing.
Regardless of how traceable the requirements, I always wonder how they were written: For example, were they written to document the intended use of the system or to impress the reviewers of the URS? Let's look at two examples of requirements, quality and traceability. Unfortunately, these are both real-life examples.
• In one periodic review, I wanted to trace a user acceptance test for the audit trail back to the URS. The URS was eventually traced to the section on "Backup and Retrieval." Requirement 5.1.3 stated "The entire system is to adhere to GXP requirements on the tracking of information. Therefore, it will be required to demonstrate the status of the system at any nominated point in time." This implies some form of audit trail is readily available. This requirement is not testable or verifiable as it does not define the actual functions of the audit trail. Interestingly, there was no mention of backup or retrieval in this section.
• Here's another example of how not to write requirements (for a spectrometer system): "6.2.4.1. Report production at least every one page every 10 s at modest network utilization." Note that the requirement is numbered, which is good for traceability. If we look at the first part of the requirement: report production of one page every 10 s, this is testable. However, then come the four words to snatch defeat from the jaws of victory: "at modest network utilization." This makes a testable requirement untestable. What is a modest network? Don't even go there! Does this mean a test is conducted at night when there are no users on the network or when they are all on a break?
These are just two examples of the rubbish requirements that the poor reviewer has to sift through.
Other Areas for Review
I do not have sufficient space to cover some other aspects of the review of the validation, such as
- Configuration of the software: Is it the same as installed or has it been changed? If yes, where is this documented and how is it tested?
- Reviewing vendor documentation critically, especially in light of my comments on vendor material (12)
- Installation documentation for the IT, instrument, and software components of the system.
Operational Review
In this section of the periodic review, we'll be looking at the different types of users, the system security (for example, look closely at the number of characters in a user's password as they log on — do the access privileges on paper match those in the system?), the procedures for the system, and use of the system in operation. Typically, you'll get out into the laboratory and talk with users and system administrators.
Depending on the scope and use of the system, this part can vary from relatively simple to very extensive, so advice here is very generic rather than detailed. Check that users' training records are current and that users have been trained on the current versions of procedures. Furthermore, do the procedures for the system describe how to use it in sufficient detail or are they vague?
A Picture Is Worth a Thousand Words
A digital camera can be very useful during a periodic review to provide supporting photographic evidence to reinforce written observations. For example, it can record an untidy or unsafe laboratory and also avoid "unofficial" corrective actions, such as tidying up the facility and then denying that a problem ever existed. If a reviewer writes in the audit report that the user name and password were written on the front of the spectrometer PC (it's a bit dry and the reader needs to imagine the situation), but if you have the section of the report as shown in Figure 3 it carries more impact. Furthermore, the reviewer can leave the reader to think, "What were those idiots thinking?" rather than leave the report to hammer the point home. You will note that the photograph in Figure 3 does not have a time and date stamp. This is deliberate because it is from an auditing course that I run, and it is one of the mistakes in an audit report that attendees have to identify.
Observation: "The user name and password were written on the front of the spectrometer PC. See the photograph below."



Figure 3: Example of photographic evidence used to support an observation in an audit report.
Regulatory Compliance — The Final Frontier?
It will be inevitable that at some stage you will need to include the IT department in a periodic review to see how activities such as backup, change, and control are carried out. Again, IT will need to be notified and have staff available according to the schedule in the plan. If the IT department activities are outsourced, then agreement with the outsourcing company may also have to be sought.
The principles here are the same as for the users: Is there a procedure, is it followed, and is there evidence of this? One issue is that some IT procedures may be written under sufferance and may be fairly basic and never read from the day they were written. However, look at the procedure to see what is written and ask the IT staff to talk about or demonstrate what is done and where the records are to demonstrate it was followed. Do they match? If yes, this is fine; but if not, then you have findings to consider.
Reviewer's Closed Meeting
The optimist in me thinks that if I allow an hour for the closed meeting, I can take my time and review what I have seen and identify the findings, and, if time allows, classify them as to their severity. The pessimist in me knows that regardless of the timetable I will overrun the review and this will reduce the time for my closed meeting. Regardless, the reviewer's closed meeting is a quiet period where audit notes and observations can be reviewed and any findings identified and classified as outlined below.
Observations, Findings, and Recommendations
What's in a word? The heading above lists observations, findings, and recommendations, but what do they mean? Again, each company will have its own definitions but these are mine:
- Observation: This is what the reviewer saw during the periodic review. Dependent on the company requirements these can be detailed or summary. However, consider that the same reviewer may not conduct the next periodic review of this system and there needs to be sufficient detail to enable another person to follow what was done in the current review. For example, a reviewer should list a standard operating procedure (SOP) with its version number so that the next reviewer can see what has changed since the last review. Note that observation as used here is not in the same context as an FDA 483 observation, it is simply what the reviewer saw during the review.
- Finding: A noncompliance with regulations or procedures equivalent to a 483 observation where there is a noncompliance with the regulations or company procedures. However, are all findings or noncompliances the same? For example, is a missing signature from a document the same as not validating a computerized system? No. Therefor,e we need to have a classification scheme. One that could be used is presented in Table IV, in which each finding is classified in ascending order of severity between 1 and 4.
- Recommendations: This is something that I am occasionally asked to do by some clients. Recommendations are not noncompliances, but simply nonbinding recommendations to the process owner by the reviewer that a working practice or way of performing a task could be improved. Whether or not the recommendation is implemented is the choice of the process owner.
The Closing Meeting
After the reviewer's closed meeting, the review is concluded by the closing meeting. Here, all involved with the audit will be present. The reviewer will lead the meeting by presenting the conclusions of the audit and the findings, and this should be done without interruption. One problem with reviews and audits is that the highest level of praise is "adequate," which means little to the people on the receiving end. The reviewer should include areas where good work has been done and say so. If there are findings, then the major ones as a minimum need to be covered. If the reviewer has been open, the laboratory staff will know where the problems are but not the classification.



Table IV: A classification of findings or noncompliances
After the reviewer has presented the initial observations and findings, the meeting is open for comment. This is important because the reviewer may have misinterpreted something that can be corrected while still on site by going through the procedures or other documented evidence. It is important for the reviewer to cover all major findings and issues directly with the laboratory staff so that there are no surprises in the report. Otherwise, they will feel aggrieved that bad news was not passed on face-to-face.
Documenting the Periodic Review
A draft report of the review is the formal outcome that will present the observations and findings, as we have discussed earlier in this column. The laboratory has an opportunity to comment on the report and feed these observations back to the reviewer. Associated with the report is a corrective action plan in which the findings (moderate and above) are listed and the laboratory is asked to complete the corrective and preventative actions against the findings. After the report and corrective action plan are agreed on, then they are finalized.
To demonstrate that a review has taken place, some organizations produce an audit certificate signed by the reviewer. This is shown to an inspector.
Summary
These two installments have discussed in some depth what is involved in a periodic review of a computerized system: the regulations, the scope of the review, and how to conduct and report one. It is the means of ensuring that computerized systems remain in control throughout their working life and must be undertaken on a regular basis.
R.D. McDowall is the principal of McDowall Consulting and the director of R.D. McDowall Limited. He is also the editor of the "Questions of Quality" column for LCGC Europe, Spectroscopy's sister magazine. Direct correspondence to: spectroscopyedit@advanstar.com



R.D. McDowall
References
(1) R.D. McDowall, Spectrosc. 26(9), 28–38 (2011).
(2) R.D. McDowall, Qual. Assur. J. 10, 15–20 (2006).
(3) M. Cahilly, presentations at the ISPE GAMP Conference, Somerset, New Jersey, 2011.
(4) US Food and Drug Administration, Current Good Manufacturing Practice regulations §211.194(a) (US Dept of Health and Human Services, Rockville, MD).
(5) EU GMP Annex 11 on Computerised Systems, (2011).
(6) PIC/S PI-011 Computerised Systems in GXP Environments, PIC/S (2004).
(7) Annex III: To Procedure For Conducting GCP Inspections Requested By The EMEA: Computer Systems.
(8) Good Automated Manufacturing Practice guidelines version 4 (International Society of Pharmaceutical Engineers, Tampa, FL, 2001).
(9) Good Automated Manufacturing Practice guidelines version 5, (International Society of Pharmaceutical Engineers, Tampa, FL, 2008).
(10) R.D. McDowall, Spectrosc. 22(11), 22–27 (2008).
(11) R.D. McDowall, Spectrosc. 22(12), 78–84 (2008).
(12) R.D. McDowall, Spectrosc. 25(9), 22–31 (2010).













