Rapid Chemical Threat Identification by SPME-GC–TMS
Special Issues
A person-portable gas chromatography–mass spectrometry (GC–MS) system employing a toroidal ion trap mass spectrometry (TMS) detector was used to analyze chemical threat related compounds. Introduction of analytes into the heated injector of the instrument was by solid-phase microextraction (SPME), and fast resistive heating of a low thermal mass (LTM) gas chromatography column assembly provided rapid analysis times. Methodology for positive identification of chemical threats can combine chromatographic retention time, comparison to traditional electron ionization mass spectral libraries, and observation of expected pseudomolecular ions produced through self-chemical ionization. Methods are discussed for sampling by SPME with GC–MS analysis in the field to measure airborne analyte concentrations.
The goal of terrorists is to create hysteria and fear using tactics that panic communities, gain publicity, impact local economies, and create political unrest (1). The deliberate release of highly dangerous chemicals including, but not limited to, chemical warfare agent (CWA) compounds could cause this type of turmoil. Systems to quickly and reliably provide information about such threats would be an important defense against this type of terrorism. Thus, a need to obtain chemical threat identification in near real-time exists to support border protection, law enforcement, first responder, and civil defense personnel.
Significant effort has gone into improving current capabilities by providing chemical detection systems with robust capabilities and optimized portability. The collection of representative gas, liquid, or solid samples followed by analysis in real-time or near real-time is an important criteria for rapid threat detection. In addition to being able to generate positive chemical threat identifications through library matching and quantification of the potential threat, the measurement system must be both easy to use and robust to have applicability in the field.
A key characteristic for high-certainty chemical analysis is orthogonality, or the ability to detect a chemical using two technologies that rely upon different chemical reactivity and chemical properties. The ability to separate chemical compounds from complex mixtures such as environmental matrices, and subsequently detect the compounds with a method having both specificity and selectivity, provides orthogonality. Historically, hyphenated methods that include a separation step (for example, gas chromatography [GC]) followed by a detector (for example, mass spectrometry [MS]) have been deployed for confirmed identification of chemical threat compounds.
GC–MS methods are used routinely with equipment located in a fixed laboratory. Van-mounted or transportable laboratory type GC–MS systems have been utilized by several organizations to address mobile response needs. These response organizations may receive only short notice for rapid mobilization. When this occurs equipment must be palletized, shipped by air, and then set up in temporary locations. The use of a transportable GC–MS system is obviously limited by the requirement to transport the vehicle to the incident site. Other person-portable GC–MS systems have been developed, but these can suffer from limited capabilities for detection of chemicals with low volatility.
To address on-site analysis needs for chemical threats, detection methods have been developed using solid-phase microextraction (SPME) (2–8) coupled with GC–MS analysis. When used in conjunction with swab, headspace, and immersion sampling protocols, SPME offers an effective sample preparation, sample concentration, and sample injection technique for a portable GC–MS system such as the person-portable GC–toroidal ion trap mass spectrometry (GC–TMS) system described by Contreras and colleagues (9) and Later and colleagues (10). When the rapid sampling capabilities of SPME are combined with a low thermal mass (LTM) GC assembly using resistive heating technology, sample analysis times in the field and the overall system size and weight can be significantly reduced compared to conventional laboratory GC systems. The resulting ability to provide near-real-time orthogonal chemical threat identification is important for personnel in the field who must make key response decisions.
In this work, SPME-GC–TMS methodology and person-portable instrumentation were used to rapidly analyze threat-related analytes known to be degradation products for the chemical warfare agent O-ethyl S-2-diisopropylaminoethyl methyl phosphonothiolate (VX).
Experimental
Chemicals: 2-(Diisopropylaminoethyl)-methyl sulfide, 2-(diisopropylaminoethyl)ethyl sulfide, 2-(diisopropylaminoethyl)isopropyl sulfide, and 2-(diisopropylaminoethyl)propyl sulfide were synthesized by reacting 2-(diisopropylamino)ethyl chloride hydrochloride (97% purity, Sigma-Aldrich, St. Louis, Missouri) with 2 equivalents of the respective thiolate sodium salt, in acetonitrile. The desired end product was extracted into pentanes along with the corresponding disulfide (for example, diethyl disulfide for synthesis of 2-(diisopropylaminoethyl)ethyl sulfide). The pentanes and volatile disulfide were then evaporated under a steady stream of Ar at 45 °C until the corresponding volatile disulfide side reaction product was no longer observed chromatographically using typical laboratory GC–MS instrumentation with liquid injections of reaction products dissolved in methanol.
Samples and Sampling: Sample introduction into the GC–TMS system used for analysis was completed through the heated injector of the instrument from an SPME fiber (65-μm PDMS/DVB coating thickness) contained in a CUSTODION SPME syringe (Torion Technologies, American Fork, Utah). Samples were prepared by injecting small aliquots of a 1-mg/mL solution of the sulfide analytes in dichloromethane into 3 L of air contained in a Tedlar sample bag. Concentrations of 0.1, 1, 10, and 100 ppm were prepared. Sampling was completed by exposing the SPME fiber to the Tedlar bag for 10, 30, 90, 270, and 710 s at room temperature.
Instrumentation: A GUARDION-7 GC–TMS instrument (Torion Technologies) described previously (9–11) was used. In the TMS instrument, ion trapping occurs at 300-Vp-p rf conditions, and mass selective instability ejection (low to high mass) is accomplished by scanning the rf voltage from 200 to 2100 Vp-p. The ejection is assisted by a 0.5–5 Vp-p, 385 MHz AC signal applied to the filament end cap.
Control of the GC column temperature is accomplished with a resistively heated LTM assembly containing a 5-m MXT-5 column (Restek, Bellefonte Pennsylvania) with 0.1 mm i.d., and 0.4 mm df. The general approach to LTM GC column module construction was described by Sloan and colleagues (12). The ruggedized LTM assembly is well suited for use in field-portable instrumentation due to its small size, weight, and low power consumption. The GC temperature program used started at 40 °C, with linear temperature ramping to 300 °C at a rate of 120 °C/min (2 °C/s). Hold times at both the lower and upper limits of the program were 10 s and 40 s, respectively. The low thermal mass injector and transfer lines from the injector to the GC column and from the column to the TMS were maintained at 270 °C. A split flow in the injector was programmed to begin 2 s following injection. The TMS m/z scan range was 41–450. The carrier gas was ultrahigh-purity (UHP) helium (He).
When considering the wide range of volatilities for analytes that can be separated and detected, and the information content provided by mass spectrometric detection, this GC–MS instrument is uniquely portable. Its overall weight is 28 lb, and its outside dimensions are small enough to allow transportation as commercial airline carry-on luggage after removing its small high pressure He carrier gas supply per airline regulations. Operation in the field using on-board batteries is possible for up to six hours of continuous use.
Results
Figure 1 is a GC–TMS total ion chromatogram (TIC) showing the rapid analysis possible using the LTM GC assembly within the GC–TMS system. The LTM GC column module used here provides for fast chromatography while greatly reducing the size, weight, and power requirements needed to rapidly attain column temperatures in excess of 300 °C. While only VX degradation products are shown here, the LTM GC system also is capable of separating VX, which is eluted late in a typical chromatographic analysis using standard laboratory GC–MS equipment. Very little tailing or other signs of activity are seen in Figure 1, demonstrating the adequacy of the passivation applied to the LTM metal injector for the analytes used here.


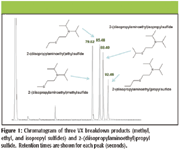
Figure 1
Mass spectra corresponding to the four GC peaks in Figure 1 are shown in Figure 2. As expected for a miniaturized ion trap mass spectrometer, mass spectra exhibit protonated molecular ions in addition to lower fragment ions which correspond well with standard 70-eV EI mass spectra available in large mass spectral libraries. The presence of [M+H]+ is in line with observations of Méchin and colleagues (13) who showed this phenomenon with ion trap detection of alkyl phosphonate compounds. The ions with m/z values greater than [M]+• are the product of interactions between trapped ions and neutral analyte molecules present concurrently in the small TMS, a phenomenon known as self-CI, as described earlier by McLuckey and colleagues (14) and others.

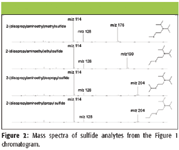
Figure 2
When using a small ion trap MS detector the ability to obtain mass spectra with similar fragmentation patterns as observed in typical reference spectra, and the ready production of spectra with pseudomolecular ion information for the same analyte in the same GC–MS analysis can increase the information available for analyte identification. However, ion–molecule interactions that produce these spectra also pose a challenge to develop data handling algorithms that will require more than just simply comparing a mass spectrum with another found in a general mass spectral database.
For quantitative applications, calibration curves using SPME-GC–TMS are possible. Note that it is important to control sampling conditions (temperature, exposure time, and so forth) when using SPME. Figure 3 shows a calibration curve for 0.1-, 1-, 10-, and 100-ppm concentrations of 2-(diisopropylaminoethyl)methyl sulfide. Similar results were obtained for the other three sulfide analytes over the same range of concentrations with correlation coefficients (R2 ) of 0.998, 0.993, and 0.998 for 2-(diisopropylaminoethyl)ethyl sulfide, 2-(diisopropyl-aminoethyl)isopropyl sulfide, and 2-(diisopropylaminoethyl)propyl sulfide, respectively.


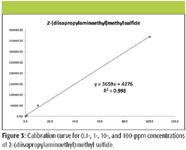
Figure 3
Under equilibrium conditions, field quantitative work can be accomplished with SPME sampling (15–17). However, to obtain quantitation under equilibrium conditions, one must know the SPME equilibrium constant (Kfs ) for each compound. The case of Kfs values for air samples can be calculated from analyte GC retention times (RTs) (15–17). A linear regression of the linear temperature program retention index (LTPRI) and RT from the table in Figure 4 were used to give equation LTPRI = 11.34RT + 335.8 with a R2 of 0.996 which was used to generate the plot of Log Kfs versus LTPRI for the VX-related analytes shown in Figure 1. The Kfs values in the table in Figure 4 were obtained from the literature and calculated as explained by Martos and colleagues (16). The LTPRI values were obtained from the NIST chromatography data base (5% phenylpolydimethylsiloxane stationary phase GC column). The retention times were generated using the same temperature program on the GC–TMS as was used for the analysis shown in Figure 1. A linear regression of the data and the plot in Figure 4 generated the equation Log Kfs = 0.004 LTPRI – 0.229. These equations were used to calculate the Kfs and LTPRI values for the sulfides shown in Table I. When equilibrium SPME sampling occurs from air with a known analyte concentration, if Kfs is known, the amount of analyte extracted (n) by the SPME coating can be determined from n = Kfs Vf C0 where Vf is the volume of the SPME phase and C0 is the initial concentration before extraction (15). This information can be used to provide a detector response factor to allow determination of analyte concentration following field analysis.

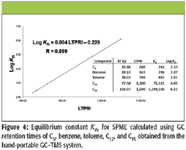
Figure 4
Ideally one would want to use the same SPME fiber as the column stationary phase or vice versa for determination of Kfs values. However this might not always be possible and usually the error due to differences in nonpolar phases is not significant. The determination of Kfs values demonstrates a method to use SPME sampling for quantitation of airborne analytes with prior analysis of a known standard and a portable GC–MS instrument.



Table I: Linear temperature programmed retention index (LTPRI) calculated from equation LTPRI = 11.34RT + 335.8 from data in Figure 4 using GC retention times shown in Figure 1 and Kfs calculated from Log Kfs = 0.004 LTPRI â 0.229 for the four sulfide compounds.
The GC–TMS system was equipped with electronic pressure control (EPC), providing two advantages. First, the mass flow through the column is constant, providing more constant and reproducible elution times, particularly for later-eluted analytes. Second, because the mass flow is constant, the vacuum conditions of the mass spectrometer remain constant. Small changes in vacuum pressure can change ion chemistry in ion trap mass spectrometers affecting the mass spectra. This will be the subject of a subsequent report.
The high portability of the GC–TMS system described, its ability to operate for hours under battery power in the field, fast chromatographic analysis speed, and orthogonal detection make it a viable option for homeland defense roles. These characteristics can allow deployment of GC–MS detection at locations where systems that produce less informative data (for example, ion mobility spectrometry detectors) typically have been considered. Thus, the instrumentation characteristics described provide the ability to deploy orthogonal detection systems at more locations than was previously possible. Such highly portable detection systems also can be deployed or moved relatively easily in response to changing threats or heightened security requirements.
Conclusions
The ability to obtain highly informative chemical threat identification data in near real-time using person-portable GC–MS instrumentation has been demonstrated using SPME-GC–TMS. With its portability and broad capabilities, the chemical detection system employing a TMS detector was used to analyze several analytes. Using fast resistive heating, LTM GC provided rapid analysis times. The production of both EI-like spectra and pseudomolecular ions in the toroidal ion trap can be used effectively for positive library identification. The addition of molecular ion information to the library search algorithm further enhances the conventional use of chromatographic retention time and traditional electron ionization mass spectral libraries. The availability of a highly portable GC–MS system with fast analysis times, extended battery power operation, and the capability to detect analytes with a wide volatility range can prove helpful for a range of homeland defense roles. This is especially important for applications in which the large size, power requirements, and relatively slow analysis speed of traditional GC–MS instrumentation would typically preclude the use of orthogonal detection. Airborne analyte concentration quantitation is possible using SPME and field-portable GC–MS.
Disclaimer
The opinions or assertions contained herein are those of the author(s) and are not to be construed as official or reflecting the views of the United States Department of Defense or the Uniformed Services University of the Health Sciences.
Edgar D. Lee, Christopher R. Bowerbank, and Douglas W. Later are with Torion Technologies Inc., American Fork, Utah. Philip A. Smith is with the Department of Preventive Medicine and Biometrics, Bethesda, Maryland.
References
(1) C. DiGiovanni, Am. J. Psych. 156, 500–1505 (1999).
(2) G.L. Kimm, G.L. Hook, and P.A. Smith, J. Chromatogr., A 971(1–2), 20, 185–191 (2002).
(3) S. Risticevic, H. Lord, T. Gorecki, C.L. Arthur, and J. Pawliszyn, Nature Protocols 5(1), 122–139 (2010).
(4) P. Smith, M.T. Sng, B. Eckenrode, S.Y. Leow, D. Koch, R. Erickson, C. Jackson Lepage, and G. Hook, J. Chromatogr., A 1067, 285–294 (2005).
(5) G. Hook, C. Jackson Lepage, S. Miller, and P. Smith, J. Sep. Sci. 27, 1017–1022 (2004).
(6) P. Smith, C. Jackson Lepage, D. Koch, H. Wyatt, B. Eckenrode, G. Hook, and G. Betsinger, Trends Anal. Chem. 23, 296–306 (2004).
(7) G. Hook, G. Kimm, G. Betsinger, P. Savage, A. Swift, T. Logan, and P. Smith, J. Sep. Sci, 26, 1091–1096 (2003).
(8) G.L. Hook, G. Kimm, P. Savage, B. Ding, and P. Smith, J. Chromatogr., A 992, 1–9 (2003).
(9) J.A. Contreras, J.A. Murray, S.E. Tolley, J.L. Oliphant, H.D. Tolley, S.A. Lammert, E.D. Lee, M. Lee, D.W. Later, and M. Lee J. Am. Soc. Mass Spectrom. 19, 1425–1434 (2008).
(10) D.W. Later, T.C. Wirth, C.R. Bowerbank, E.D. Lee, and C.S. Sadowski, Spectroscopy 27(4S), 8–15 (2009).
(11) S.A. Lammert, A.A. Rockwood, M. Wang, M. Lee, E.D. Lee, S.E. Tolley, J.L. Oliphant, J.L. Jones, and R.W. Waite, J. Am. Soc. Mass Spectrom. 17, 916–922 (2006).
(12) K.M. Sloan, R.V. Mustacich, and B.A. Eckenrode, Field Anal. Chem. Technol. 5, 288–301 (2001).
(13) N. Méchin, J. Plomley, R.E. March, T. Blasco, and J.-C. Tabet, R. Commun. Mass Spectrom. 9, 5–8 (1995).
(14) S.A. McLuckey, G.L. Glish, K.G Asano, and G.J. Van Berkel, Anal. Chem. 60, 2312–2314 (1988).
(15) G. Ouyang and J. Pawliszyn, Anal. Bioanal. Chem. 386, 1059–1073 (2006).
(16) P.A. Martos, A. Saraullo, and J. Pawliszyn, Anal. Chem. 69,402–408 (1997).
(17) P.A. Martos and J. Pawliszyn, Anal. Chem. 69, 206–221 (1997).















Mass Spectrometry for Forensic Analysis: An Interview with Glen Jackson
November 27th 2024As part of “The Future of Forensic Analysis” content series, Spectroscopy sat down with Glen P. Jackson of West Virginia University to talk about the historical development of mass spectrometry in forensic analysis.

Detecting Cancer Biomarkers in Canines: An Interview with Landulfo Silveira Jr.
November 5th 2024Spectroscopy sat down with Landulfo Silveira Jr. of Universidade Anhembi Morumbi-UAM and Center for Innovation, Technology and Education-CITÉ (São Paulo, Brazil) to talk about his team’s latest research using Raman spectroscopy to detect biomarkers of cancer in canine sera.



