The Role of Mass Spectrometry in Biopharmaceutical Drug Discovery and Development
Spectroscopy
As biopharmaceuticals continue to increase in sophistication, how can their discovery and development be managed to reduce uncertainties and expedite the process?
The discovery and development of biopharmaceuticals that target specific diseases can be transformative for people living with illness. However, bringing a new therapy to market is a prolonged and costly process mired in uncertainty. Ensuring safety, efficacy, and product quality is paramount. Biopharmaceuticals, by their nature, are highly complex. A myriad of heterogeneity can be intentionally functional, an unwanted consequence of manufacturing and storage, or generated by biological modification in vivo. Not all, but some post-translational modifications or biotransformations can impact development, manufacturing, safety, efficacy, and overall product quality. These critical quality attributes (CQAs) need to be identified, characterized, controlled, and monitored throughout the drug discovery and development cycle. Specialty measurement using mass spectrometry (MS) continues to play an ever-increasing role across the continuum.
The biopharmaceutical universe can be mind boggling at times-a menagerie of shapes and sizes, spanning a vast and expanding array of complexity including: oligonucleotides, peptides, synthetic peptides, therapeutic proteins, monoclonal antibodies (mAbs), fusion proteins, bispecific antibodies, antibody–drug conjugates (ADCs), and CAR-T cells (1).
For example, the Scripps Research Institute, in collaboration with Janssen Research & Development, recently announced the development of a synthetic cyclic peptide mimicking the binding region of a neutralizing antibody that offers broad spectrum immunization to influenza including the avian influenza strain H5N1 (2). In contrast to therapeutic antibodies, peptides can be easier to develop, manufacture, and deliver to patients. For rare diseases, like short bowel syndrome, the U.S. Food and Drug Administration (FDA) can grant orphan drug status for fast track development, such as Zealand Pharma's glepaglutide peptide.
Other examples include the many therapies combating immunology and oncology diseases that are in development. A recent Science publication by a public-private partnership describes a trispecific antibody with broad neutralizing capability to HIV that is being produced by Sanofi to be used in a phase I clinical trial (3). Recently, the FDA approved Novartis's first-in-class CAR-T cell therapy, which reengineers a patient's own T cells, using gene transfer technology, to recognize specific antigens on the surface of leukemia tumor cells initiating cell death (4). Modalities such as these are leading the march towards personalized medicine.
Managing Uncertainty
How is the discovery and development of a complex biopharmaceutical managed to reduce uncertainty in approval and what can be done to expedite the process? In 2014, AstraZeneca published a landmark R&D productivity retrospective, noting the highest rate of drug failure, industry wide, was in phase II clinical trials, with a large percentage of projects failing because of poor safety and efficacy. This catalyzed a new "five Rs" strategy to guide research and development and requires the right target, tissue, patient, safety, and commercial potential (5). Emphasis is placed from the start of the product development life cycle on understanding the disease mechanism and the drug mechanism of action or toxicity. As a consequence, drug development now includes efficacy biomarkers that can report target engagement at an earlier stage. To reduce uncertainty in efficacy downstream, recruitment to clinical trials now incorporates patient stratification, including larger populations, often with genetic linkage to the disease in question.
A target product profile (TPP) that summarizes the drug development program in terms of labelling concepts is designed to encourage dialogue between sponsor and regulator, reduce risk of late-stage drug development failures, and ensure that safety and efficacy data is available in a timely manner. As stated in the FDA guidance, the TPP embodies the notion of beginning with the goal in mind (6). The TPP contains a description of product attributes (PAs) including physical and chemical characteristics, such as amino acid sequence and post-translational modifications (PTMs). Characterization starts in late-stage discovery, prior to candidate selection for clinical trials. In early development, the PAs need to be assessed for impact on bioactivity, safety, pharmacokinetics (PK)–pharmacodynamics (PD), and immunogenicity risk, and flagged as potential critical quality attributes (pCQAs) (7).
The International Society for Cellular Therapy (ISCT) hosts annual meetings between the FDA's Center for Biologics Evaluation and Research (CBER) and stakeholder organizations to discuss specific concerns arising in the field of biopharmaceutical drug development. On the radar at its last meeting in 2016, as presented by Tom Finn, a product reviewer from the CBER's Office of Tissues and Advanced Therapies, was the impetus to establish and apply critical quality attributes (CQAs) during a product's development life cycle-with a focus on those characteristics ensuring safety and efficacy (8).


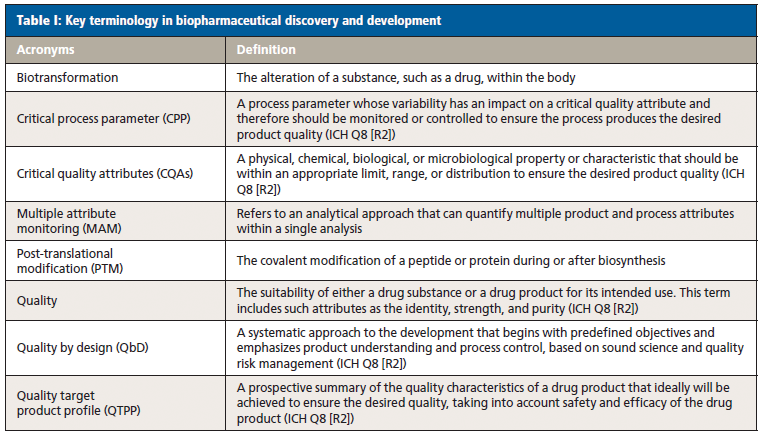
PTMs or biotransformations that affect safety and efficacy, for example methionine oxidation in the complementarity-determining region of mAbs influencing antigen binding and potency, are classified as CQAs. Analytical methods that enable identification, characterization, control, and monitoring of CQAs with high certainty are the glue that binds a drug discovery and development program together. The TPP mentioned previously can change throughout the product development life cycle evolving into the quality target product profile (QTPP). There is significant overlap in managing CQAs during pilot manufacturing and preclinical testing. A PTM or in vivo biotransformation that surfaces in preclinical studies and which affects stability or efficacy may require product or process optimization, and be recognized as a CQA in manufacturing process development. In turn, multiple CQAs may need monitoring downstream in quality control (QC).
Application of Mass Spectrometry in Discovery and Development
A biopharmaceutical product can consist of variants or subspecies derived from functional PTMs or modifications, including, for example, glycosylation variants (mAbs) or ADCs. Techniques that can characterize and quantify this level of complexity are in high demand. Liquid chromatography–mass spectrometry (LC–MS) is well-established for biopharmaceutical characterization because it has the ability to make direct qualitative and quantitative measurements at the molecular level. However, usage is growing in other areas including large molecule drug metabolism pharmacokinetics (DMPK), process development, manufacturing, quality control, aftermarket surveillance, and therapeutic drug monitoring. For discovery research, new and advanced LC–MS technology also exists (9).
High resolution mass spectrometers (HRMS), particularly quadrupole time-of-flight mass spectrometers (QTOFs), are well-established for biopharmaceutical characterization in drug discovery and development. The inherent capability of TOF mass spectrometry for high fidelity detection, for example, enables accurate glycosylation profiling of mAbs (10), or quantification of drug antibody ratio (DAR) in ADCs-both of which can be important CQAs (11).
Modern high-end QTOFs have the mass resolution, mass accuracy, and sensitivity-in many cases exceeding the requirements-for routine applications like peptide mapping, intact protein analysis, and glycosylation profiling (12). The ability to obtain information quickly and having high confidence in data integrity is just as important as general instrument performance. As is the industry's need to manage data efficiently and easily, including transfer of information across life cycle stages, while interfacing with regulated and nonregulated stakeholders-including strategic partners.
Application of Mass Spectrometry in Pharmacokinetics
In PK studies, drug clearance needs to be measured quantitatively. Traditionally, drug concentration is measured post-dose using ligand bind assays (LBA). However, LBAs can be limited by poor specificity and cross-reactivity. Depending on the type of reagents used, LBAs can be insensitive to protein isoforms and variants. Changes in biopharmaceutical structural integrity via biotransformation can cause reagent-ligand binding issues, and anti-drug antibodies elicited via immunogenicity can lead to an underestimation of true concentration (13). Alternative techniques with greater specificity and multiplexing capability such as MS are being used more frequently in nonregulated bioanalysis to support regulatory submissions, offering "fit for purpose" sensitivity and dynamic range with the advantage of direct quantitation for greater confidence and reduced measurement uncertainty.
Tandem quadrupole mass spectrometry (TQMS) using the surrogate peptide approach is still the gold standard for protein quantification when the highest levels of sensitivity, accuracy, and precision are required (14,15). Validated assays can be performed routinely, and the technology is well-suited to large-scale studies and therapeutic drug monitoring (16,17). However, quantification can also be performed by HRMS, and there is growing interest in taking advantage of high mass resolution and accurate mass when sensitivity is hampered by inadequate selectivity (18). HRMS is also attractive for method development. Data independent acquisition or targeted MS/MS enables all peptide fragments to be detected at high mass resolution for selection of "high specificity" transitions that can be transferred to a higher sensitivity mode such as TOF-multiple reaction monitoring (MRM), or TQMS for routine analysis. The number of molecules allowed into the analyzer is not limited with a quadrupole time-of-flight mass spectrometer compared to ion trap technology and is therefore especially attractive for quantification of biotherapeutics in complex matrices with a high background at the lower limit of quantification (LLOQ) (19).
Quantification using the surrogate peptide approach requires complete protein digestion and careful selection of unique peptides that can report on total protein or protein variants of interest (20). However, uncertainty can arise if the chosen surrogate peptides are blind to biotransformations including endogenous proteolytic cleavage or partial protein degradation. In contrast, intact protein analysis, without digestion, retains molecular integrity and is attractive for quantification of the protein pool as a whole. Compared to TQMS, proteins exceeding ~10–15 kDa can be analyzed at the intact level by HRMS including isoforms (for example, monoclonal antibody glycoforms, Δ 162 Da). In complex matrices like plasma, immunocapture is also an option. Partial digestion of protein into subunits can also be performed, retaining primary structure and enabling whole molecule quantification, including glycoforms or analysis of ADC biotransformation (for example, payload hydrolysis, Δ 18 Da) (21,22). For PTMs or biotransformations that result in smaller mass changes such as deamidation (+ 1 Da) the protein pool can be fully digested and the different subspecies quantified at the surrogate peptide level (20). In addition, intact protein analysis can be used to measure large biotransformation-related changes, for example truncation and catabolism in fusion proteins caused by unknown proteolytic cleavage in linkers.
HRMS is well-established for small molecule metabolite identification, and is increasingly being sought by DMPK scientists for large-molecule catabolism and biotransformation analysis. This demand requires informatics workflows to track and quantify clearance of peptide and protein biotherapeutics, and to understand biotransformation events, particularly for molecules that include non-natural amino acids and linkers that affect in vivo stability, bioactivity, and safety (23,24).
Application of Mass Spectrometry in Development
Maintaining the chemical and structural integrity of therapeutic antibodies during manufacturing, distribution, and storage remains a major challenge for pharmaceutical development. Establishing protein structure–function relationships, identifying CQAs and CPPs, and developing an effective control strategy are therefore important throughout drug development (7).
One example that illustrates this point is methionine oxidation, which is a commonly observed modification in protein therapeutics. In a recent publication, Janssen scientists utilized a forced degradation study, hydrogen-deuterium exchange (HDX) HRMS, and structural modeling, and successfully differentiated the impact of oxidation at individual methionine residues, providing highly valuable information to assist CQA assessment of antibody therapeutics (25).
Once CQAs are identified, developing an effective control strategy is essential to ensure successful commercial manufacturing of high-quality medicines. High-throughput, in-line, or at-line analytical tools are critical to establish the correlations between process parameters and product quality attributes to help advance process understanding, improve product quality, and increase production efficiency (26).
In addition to identifying CQAs and CPPs, robust analytical methods should also be developed and validated for QC purposes to prepare for commercial launch. As a result of its unique capabilities of multiattribute monitoring (MAM), HRMS could have tremendous potential in quality control. Recently, Janssen scientists have demonstrated a quantitative MS-based subunit methionine oxidation analysis method that is sensitive and robust and provides much higher throughput than the traditional peptide mapping analysis (27).
Quality by design (QbD) in development identifies CQAs relevant to product quality, safety, and efficacy. Understanding their relationship to bioactivity, PK–PD, immunogenicity, and stability will guide a control strategy. Numerous CQAs including PTMs (isomerization, oxidation, deamidation, and glycosylation) need to be monitored. The CQAs may have been characterized in early development using research-grade HRMS, however, in manufacturing, the same attributes need to be monitored and quantified in production using low-cost and easier to operate mass detectors that can be dependably deployed in a QC environment. A team at MedImmune reported recently a "multi-attribute monitoring method using a quadrupole dalton mass detector to selectively monitor and quantitate PTMs in a therapeutic monoclonal antibody," which was qualified according to the International Conference on Harmonization (ICH) guidelines and applied to product characterization and stability (28).
Conclusions
The increasing complexity of biopharmaceuticals and the evolving demand for powerful bioanalytical strategies including LC–MS to characterize, quantify, control, and monitor them gives rise to a continuous "parlay" between exploiting analytical data reporting on safety, efficacy, and product quality, and issuance of guidance that enables acceptance by regulatory bodies for evaluation of risks and benefits and approval of revolutionary drugs. One thing is clear, biopharmaceuticals have continued to increase in sophistication in an era of personalized therapies, and innovation in LC–MS and informatics will continue to meet industry demands (29).
References
(1) https://www.amgenscience.com/the-shape-of-drugs-to-come/.
(2) https://www.scripps.edu/news/press/2017/20170928KadamWilson.html.
(3) L. Xu et al., Science 358(6359), 85–90 (2017).
(5) D. Cook et al., Nature Reviews Drug Discovery 13, 419–431 (2014).
(6) https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm080593.pdf.
(7) N. Alt, Biologicals 44, 291–305 (2016).
(8) Establishing and Applying Critical Quality Attributes During the Product Development Lifecycle, ISCT Liaison Meeting, 19 October 2016, Tom Finn, Product Reviewer (FDA, CBER).
(9) M.A Moseley et al., J. Proteome Res. Sep 13, Epub ahead of print (2017).
(10) V. D'atri, Anal. Chem. 89, 2086-2092 (2017).
(11) E.A Redman et al., Anal. Chem. 88, 2220–2226 (2016).
(12) A. Beck et al., J. Mass Spetrom. 50, 285–297 (2015).
(13) M. Kelley et al., The AAPS Journal 15(3), 646–658 (2013).
(14) R.J. Mbasu et al., Proteomics 16(15–16), 2206–2220 (2016).
(15) A.J. Percy et al., EuPA Open Proteomics 8, 6–15 (2015).
(16) Y. Xu et al., J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 1063, 50–59 (2017).
(17) X. Dong et al., J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 1063, 204–213 (2017).
(18) J.F. Kellie et al., Bioanalysis 8(3), 169–177 (2016).
(19) A. Kaufmann and S. Walker, Rapid Commun. Mass Spectrom. 30(8), 1087–95 (2016).
(20) P. Bults et al., Anal Chem. 88(3), 1871 (2016).
(21) J.F. Kellie et al., Bioanalysis 8(20), 2103–2114 (2016).
(22) B. Rago et al., Bioanalysis 8(21), 2205–2217 (2016).
(23) M.P Hall, Drug Metab. Dispos. 42, 1873–1880 (2014).
(24) Mark Wrona et al., White Paper: Biopharmaceutical System Solution with UNIFI Scientific Information System for the Evaluation of Peptide Catabolism, Waters Corporation, Milford, Massachusetts.
(25) J. Mo et al., Anal Chem. 88, 9495-9502 (2016).
(26) J. Dong et al., Anal. Chem. 88, 8673–8679 (2016).
(27) I. Sokolowska et al., mAbs 9(3), 498–505 (2017).
(28) W. Xu et al., mAbs 1–11 (2017).
(29) G.T. Roman, Bioanalysis 9(14), 1033–1035 (2017).
Ian John Edwards is a pharmaceuticals business development manager at Waters Corporation.


















Chinese Researchers Develop Dual-Channel Probe for Biothiol Detection
April 28th 2025Researchers at Qiqihar Medical University have developed a dual-channel fluorescent probe, PYL-NBD, that enables highly sensitive, rapid, and selective detection of biothiols in food, pharmaceuticals, and living organisms.
.")


