The Use of Mass-Spectrometry-Based Proteomics for Reliable Detection and Identification of Pathogens as Illustrated by the Study of Bee Colony Collapse Disorder
Special Issues
A study of colony collapse disorder in honey bees illustrates how mass spectrometry–based proteomics techniques can be used to to identify pathogens without any prior knowledge of what is contained in the sample.
Identification and classification of pathogenic microorganisms, particularly of pre-existing or emerging strains of infectious agents such as bacteria or viruses, is an important concern. The main shortcoming of most current methods, which rely on reverse transcription polymerase chain reaction, is that they require known sequence information for detection. Mass spectrometry–based proteomics (MSP) techniques, in contrast, can be used to identify pathogens without any prior knowledge of what is contained in the sample. MSP also can be done without the need to grow cultures, and enable the microorganism to be classified down to strain level. A study of colony collapse disorder in honey bees illustrates these techniques.
Methods based on reverse transcription polymerase chain reaction (RT-PCR) are used extensively in the diagnosis of genetic diseases as well as in the determination of the abundance of RNA molecules within a cell or tissue as a measure of gene expression. However, RT-PCR requires known sequence information to reliably detect and identify pathogenic microorganisms. For example, the technique uses probes that hybridize to a known sequence. If a significant mutation occurs in a viral target, the known effective probes are rendered useless, because they will be unable to hybridize to a mutant sequence.
By using mass spectrometry (MS)–based proteomics (MSP) techniques, along with rigorous sample processing methods, it becomes possible to effectively identify and quantify thousands of proteins in both healthy and infected samples in a single study. MSP offers an orthogonal and complementary approach to the RT-PCR that has been traditionally used for pathogen identification and classification. The technology is capable of generating unambiguous peptide fragment data that are then processed using bioinformatics tools in which the fragments are compared to a full library of peptide sequences generated from genomics and proteomics research. As a consequence, peptide fragment data acquired by MSP enables the identification and classification of microorganisms without the need for amplification, probes, or primers that are commonly associated with RT-PCR–based methods. It is also of significant importance that MS allows for the identification, quantification, and classification of fungi, bacteria, and viruses in a single analytical run. Phylogenetic classification can be to strain level and is limited only by the level of precision within the proteomic and genomic databases.
In a recent publication (1), we presented the findings of a study whereby the MSP approach was deployed to identify and quantify thousands of proteins from healthy and collapsing bee colonies. Colony collapse disorder (CCD) is a major cause of concern in the United States because it results in the devastation of numerous honey bee colonies every year. To date, many CCD investigations, using sensitive genome-based methods such as RT-PCR, have identified small RNA bee viruses and the microsporidia Nosema apis and Nosema ceranae in both healthy and collapsing colonies. Nevertheless, the use of these techniques has not yet made it possible to firmly link any pathogen to honey bee losses.
Experimental
More than 60 different bee samples were analyzed for pathogens using an MSP technique. One of the samples originated from U.S. honey bee colonies that had been affected by CCD in 2006–2007, when this unusual syndrome first received worldwide press coverage. Another sample was from a collapsing colony in an observation hive fitted with a bidirectional flight counter and was sampled through time as the colony failed. Another was collected from collapsed bee colonies in Florida. Non-CCD Australian honey bees were used as reference sample. A further sample came from an isolated nonmigratory beekeeping operation in Montana with no history of CCD and another consisted of dead bees recovered from inoculation feeding trials with N. ceranae alone, invertebrae iridescent virus (IIV) alone, a mixture comprising N. ceranae plus IIV, and controls that were fed syrup alone.
The paper describes the sampling methods, inoculation experiments, and MSP protocols that were used during the study (1). The experimental MS-MS spectral data of bacterial peptides were searched using the SEQUEST algorithm (University of Washington) against a constructed proteome database of microorganisms.
Results
Following MSP analysis of all these samples by ABoid (U.S. Army) (2), a database of more than 3000 identifiable peptides was generated, which represented more than 900 different species of invertebrate-associated microbes. Of those, only 29 were identified as being specific to bees and were used for subsequent analyses. The study focused on viruses, fungi, and microsporidia in the genus Nosema.
MSP analysis detected two RNA viruses (Varroa destructor-1 virus and Kakugo virus) that had not been previously reported in North American honey bee populations. A highly significant and also unreported co-occurrence of strains of DNA of IIV was also identified, featuring a microsporidian of the genus Nosema. The two RNA viruses were only noted occasionally, but the DNA virus was present in virtually all CCD samples. IIV were identified as the most prevalent viral peptides, appearing with 100% frequency and at high peptide counts in failing and collapsed colonies. They were also present in almost 75% of strong colonies, although at lower concentrations.
Invertebrae iridescent viruses are large double-stranded DNA viruses of the Iridoviridae family, and their potential correlation with CCD had not been noted previously because small RNA viruses have been traditionally associated with the occurrence of most bee diseases. As a result, the detection and identification of IIV constitutes a very important finding and a step forward toward solving the CCD problem. The study suggests that the interaction between N. Cerenae and an IIV-6-like virus could contribute to bee mortality. Furthermore, it was noted that IIV and N. ceranae occurred throughout a collapse and their concentrations increased as the severity of the CCD increased.
Inoculation cage-trial experiments were subsequently used to test the MSP-generated hypothesis that an interaction between N. ceranae and IIV may lead to increased bee mortality. The results of these experiments are shown in Figure 1.


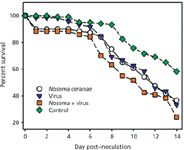
Figure 1: Survival over a 14-day postinfection period observed in cage-trials of honey bees infected with Nosema ceranae and IIV. Figure represents the combined survival results for four biological replications; N = 30 bees in each group for each biological replicate (1).
The figure plots the survival over a 14-day post-infection period observed in cage-trials of newly emerged, 1–3 day old honey bees infected with N. ceranae and IIV. The figure represents the combined survival results for four biological replications (N = 30 bees in each group for each biological replicate). Deaths in control group were confirmed not to be pathogen-related via MS analyses. The survival data were analyzed using the Kaplan-Meier method, with the following results: control vs. N. ceranae: P = 0.01; control vs. IIV alone: P,0.01 (0.008); control vs. Nosema + IIV: P,0.01 (0.0001); Nosema alone vs. IIV alone: P = 0.90; Nosema alone vs. Nosema + IIV: P = 0.04; virus alone vs. Nosema + IIV: P = 0.04. (Bees that perished within 24 h of inoculation were not included in the survival curve analyses.) These results strongly supported the hypothesis that coinfection with N. ceranae and IIV can be more lethal to bees than either pathogen alone.



Figure 2: Discriminant function analysis for differences in pathogen peptide counts among strong, failing, and collapsed honey bee colonies. Vertical and horizontal lines mark the non-CCD out-group as a reference set (1).
Stepwise discriminate function analysis of count-weighted occurrence data was undertaken to assess whether strong, failing, and collapsed colonies could be differentiated by specific patterns of pathogen occurrence (Figure 2). In the figure, function 1 explains 81% of the discriminating variance and contrasts a higher incidence of iridovirus (IIV), Nosema, and to a lesser extent, black queen cell virus (BQCV) in failing colonies with a higher incidence of deformed wing virus (DWV) and some Israel acute paralysis virus (IAPV) in the remaining groups. Thus, only two pathogens, namely IIV and DWV, were necessary for significant discrimination among different colonies. A noteworthy finding was that collapsed and strong colonies were not significantly different; however, this finding can be easily explained as the few bees left in colonies at the final stages of collapse are those that are not infected.
The SEQUEST algorithm was implemented to search the experimental MS-MS spectral data of bacterial peptides against the proteome database of microorganisms. SEQUEST thresholds for searching the product ion mass spectra of peptides were Xcorr, deltaCn, Sp, RSp, and deltaMpep. These parameters provided a uniform matching score of all candidate peptides, the generated outfiles of which were subsequently validated using a peptide prophet algorithm (3,4).
Conclusion
The mass spectrometry–based proteomics approach is set to play a key role in the life science, medical research, environmental, and food testing markets over the next few years. As the recent use of this method in the study of honey bee colony collapse disorder illustrates, MSP provides an unrestricted and unbiased approach for sensitive and cost-effective detection and identification of pathogenic microorganisms down to strain level.
The major differentiating advantage of MSP is that it is able to identify bacteria, viruses, fungi, and other cellular material without the need for growing cultures or any prior knowledge about them. In addition, MSP is faster than PCR-based methods. The entire MSP process (sample preparation, mass spectrometry, and software processing) takes about an hour to complete, depending on the complexity of the sample. In addition, the computer file can be analyzed again when the genomic data are updated and in this manner identify previously unknown microbes at a later time without having to re-examine the samples. This method is of significant benefit for infectious disease identification and a range of other potential applications in military, medical, pharmaceutical, food, and public safety areas.
Author's Note
The data discussed here were previously published in reference 1.
Charles H. Wick is retired from his position as a senior research scientist at the US Army Edgewood Chemical Biological Center, in Aberdeen Proving Ground, Maryland. He can be reached at charles.h.wick@us.army.mil.
References
(1) J.J. Bromenshenk et al., PLoS One 5(10) (2010): e13181. doi:10.1371/journal.pone.0013181.
(2) S.V. Deshpande et al., J. Chromatogr. Separat. Techniq. S5:001. doi:10.4172/2157-7064. S5-001 (2011).
(2) R. Aebersold and M. Mann, Nature 13, 198–207 (2003).
(3) A. Keller, A. Nesvizhskii, E. Kolker, and R. Aebersold, Anal. Chem. 74, 5383–5392 (2002).















Mass Spectrometry for Forensic Analysis: An Interview with Glen Jackson
November 27th 2024As part of “The Future of Forensic Analysis” content series, Spectroscopy sat down with Glen P. Jackson of West Virginia University to talk about the historical development of mass spectrometry in forensic analysis.

Detecting Cancer Biomarkers in Canines: An Interview with Landulfo Silveira Jr.
November 5th 2024Spectroscopy sat down with Landulfo Silveira Jr. of Universidade Anhembi Morumbi-UAM and Center for Innovation, Technology and Education-CITÉ (São Paulo, Brazil) to talk about his team’s latest research using Raman spectroscopy to detect biomarkers of cancer in canine sera.



