Validation of Spectrometry Software: Risk Analysis Methodologies for Commercial Spectrometer Software
A simplified risk analysis methodology is needed for the validation of commercial software used in the regulated laboratory. In this column, the author takes a look at two alternative approaches.



In the last "Focus on Quality" column (1), I noted that the risk analysis methodology used in the GAMP Good Practices Guide for Validation of Laboratory Computerized Systems (GPG) (2), which was based upon failure mode effect analysis (FMEA), was too complicated. The rationale was that we purchase mostly commercial software in regulated laboratories that already has been tested by the respective vendor. Therefore, why do we need to potentially repeat work that has already been performed?
Before we begin this discussion, it is worth mentioning that I have written a paper on risk assessment at the system level, and determination of how much validation is required has been published recently by Advanstar Communications (3). This publication looked at the nature of the software used in the system and the impact of the records produced by the system to determine how much validation was required. The outcome if validation was required was one of two options: Full validation based upon a system implementation life cycle when the detailed steps to be undertaken are defined in a validation plan for the system. Lower-risk systems could be validated using a single integrated validation document that defined the intended use, security and access control, compliance issues, and preservation of the records produced by the system.
In this column, I want to go into more detail and compare the FMEA used by the GPG with a simpler and, in my view, quicker risk analysis methodology called functional risk assessment (FRA).
Document Your User Requirements
Before beginning the discussion, what should be our starting point for the risk analysis and risk assessment?To be effective, any risk analysis is based upon available information (4). Therefore, the best source of this is the user requirements specification (URS). When you are conducting a risk assessment at the requirements or functional level, the URS should be relatively complete. This means that you have a good understanding of what the system will be doing, and this is reflected in the quality of the user requirements. If you don't have this, there is little point in going further, as you will be wasting your time regardless of which risk methodology you select.
Modified FMEA
The risk assessment methodology approach flow chart used in the GPG is shown in Figure 1. It is a two-phase approach that determines first the system risk, then at the functional level the hazards, the probability of occurrence, and their severity to identify and prioritize the risks that could occur when using the system.


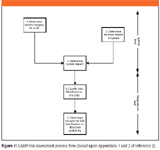
Figure 1: GAMP risk assessment process flow (based upon Appendices 1 and 2 of reference 2).
The process works as follows and is divided between assessing the overall system impact and testing priority of the hazards of the individual functions that comprise it:
1. Classify the system. This uses the seven different categories between A and G as we discussed in the previous column (1) and assesses the technical complexity of a system.
2. Next, the business impact of the system is determined. This is a plot of the compliance risk (assessed as low, medium, or high) versus business risk based on the importance of the system to the business (also low, medium, and high). This determines the business impact as a single figure between 1 and 5 (a range from very low to very high), shown in Figure 2.



Figure 2: Determination of business impact of a system (adapted from reference 2).
Compliance risk can be laboratory issues such as data integrity and traceability of calculations or the wider impact of patient safety. Business risk can be items such as data loss, inability to release a batch, or in a wider context, a product recall. The criteria that are used for both estimations will have to be developed by the laboratory, possibly in conjunction with a validation or quality assurance group.
3. The overall system impact is determined by plotting the system category (A–G) versus business impact (1–5) to produce a 7 3 5 grid to determine the overall system impact between 1 and 3 (low, medium, and high impact) as shown in Figure 3 as a traffic light scheme from green to red. The grid used in this assessment is relatively large; furthermore, as we saw in the last column, a system can be classified in two or three system classes. This means that if you misclassify a system, there is the potential to either over- or underrate the system impact.



Figure 3: Determination of overall system impact (adapted from reference 2).
4. Having determined the overall system impact, the attention now turns to the criticality of individual requirements and the hazards that a system faces. Conceptually, it is better to focus on the criticality of requirements rather than the hazards because we are dealing with off-the-shelf or configurable commercial software. The main difference is that when you write a macro or custom software for use with your spectrometer software, you may need to consider the hazards that the macro is faced with, as you will be dealing with a unique situation with no help available from the vendor. Either way, the requirement of the hazard is classified as low, medium, or high.
Now you need to consider the risk likelihood. This is fine for a hazard where the likelihood can be seen as low, medium, or high as "the frequency of an adverse event" and can be estimated for a specific hazard relatively easily. Plotting the likelihood versus effect of the hazard allows the risk to be classified as level 1, 2, or 3 as shown in Figure 4. A level 1 risk is the highest, as shown by the red coloration, and a level 3 risk is the lowest, shown as green.


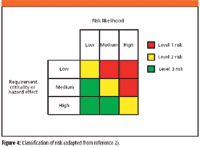
Figure 4: Classification of risk (adapted from reference 2).
Now here comes a major problem—if we are using criticality of requirement, then how is this linked to risk likelihood? I have no idea, because there is nothing in Appendix 2 of the GPG that describes this situation (2).
5. Finally, the ability of the system to detect the risk occurring is estimated as low, medium, or high and plotted against the risk level to determine the highest to lowest priority for testing as shown in Figure 5.



Figure 5: Determination of testing priority (adapted from reference 2).
The biggest issue I have with this approach is that the intended use of the system has been relegated to a sideshow with the overriding emphasis placed on risk assessment. We must not forget that we have other control mechanisms in place in the laboratory — maintenance and calibration — and these are key ways to ensure the overall quality of data and information generated by a spectrometer.


Figure 6: Functional risk assessment process flow chart.
Functional Risk Assessment
FRA is a simpler risk analysis methodology that was developed specifically for the validation of commercially available software. The process flow in Figure 5 is described in the list below, and the numbers in the figure correspond to each task described below.
1. The input to the process is a prioritized user requirements specification. All URS requirements are prioritized as either mandatory (M) or desirable (D). The mandatory assignment means that the requirement must be present for the system to operate; if "desirable" is assigned, then the requirement need not be present for operability of the system — it is simply nice to have (5,6). This is shown in Table I with the first three columns.


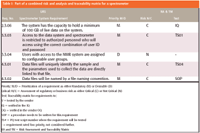
Table I: Part of a combined risk and analysis and traceability matrix for a spectrometer
Each requirement is numbered, specified, and prioritized and is the format of a URS used in this methodology.
2. The next stage in the process is to carry out a risk assessment of each function to determine if the function is business or regulatory risk critical (C) or not (N). This risk analysis methodology uses the tables from the URS that have two additional columns (columns 4 and 5) added to them as shown in Table I. The approach is shown in Table I in the fourth column from the left. Here, each requirement has been assessed as either critical or noncritical. For a requirement to be assessed as critical, one or both of the following criteria need to be met.
The requirement functionality poses a regulatory risk that needs to be managed. The basic question to ask here is: will there be a regulatory citation if nothing is done? For example, requirements covering security and access control, data acquisition, data storage, calculation and transformation of data, use of electronic signatures, and integrity of data are areas that would come under the banner of critical regulatory risk (as well as good science). Requirements can also be critical for business reasons — for example, performance of the system or its availability.
3. The functional risk assessment approach is based upon plotting the prioritized user requirements and regulatory or business criticality together to produce the Boston Grid shown in Figure 3. Requirements that are both mandatory and critical are the highest risk (combination of the prioritization and business–regulatory risk).
For most commercial spectrometry systems, requirements either fall into the high- or the low-risk categories. There will be a few requirements in the mandatory and noncritical quadrant of the grid, but few, if any, in the desirable but critical quadrant. This is logical. If your requirement were only desirable, why would it be critical or vice versa? If requirements fall in this last quadrant, it may be an indication that the initial prioritization or the risk analysis was wrong, and either should be reassessed.
4. Under the FRA, only the software requirements classified as "high" in the grid (mandatory and critical) will be considered further in the validation of a system. No other combination (that is, low) will be considered any further.
5. Once the risk analysis has been completed, the advantage of the FRA approach is that a traceability matrix can be included in the same document. This is achieved by adding a fifth column to highlight where the requirement will be tested or verified in the remaining part of the validation. Some of the areas are illustrated in the legend to Table I and also are shown in the table itself.
Limitations of FRA
The underlying assumption of FRA is that the software (together with the instrument) has undergone more testing by the vendor during development than anybody in a laboratory can undertake in a reasonable timeframe. Moreover, anticipated problems with the software or the records have been considered and detailed designs implemented and tested to cope with these situations. Therefore, using this premise as a basis, it sits on top of a vendor's approach. However, this assumption should be verified by a vendor audit for critical systems.
The application of FRA is intended for use with commercial off-the-shelf (COTS) software (GAMP Category 3) and configurable COTS software (GAMP category 4). It has not been applied to bespoke or custom-coded systems (GAMP category 5).
Summary
Two risk analysis methodologies for validation of computerized laboratory systems are compared and contrasted: a modified FMEA and FRA. The latter is a simpler methodology that is easy to understand and apply, plus it has an advantage that the traceability matrix can be combined relatively easily and with little effort.
R.D. McDowall is principal of McDowall Consulting and director of R.D. McDowall Limited,and "Questions of Quality" column editor for LCGC Europe, Spectroscopy's sister magazine. Address correspondence to him at 73 Murray Avenue, Bromley, Kent, BR1 3DJ, UK.
References
(1) R.D. McDowall, Spectroscopy 21(4), 14–30 (2006).
(2) GAMP Good Practice Guide on the Validation of Laboratory Computerized Systems (ISPE Tampa FL, 2005).
(3) R.D. McDowall, Pharmaceutical Regulatory Compliance Guidebook, In Press.
(4) ISO 14971: Risk Management for Medical Devices, Section 2 Definitions (International Organization for Standardization, Geneva, 2000).
(5) R.D. McDowall, Validation of Chromatography Data Systems: Meeting Business and Regulatory Requirements (Royal Society of Chemistry, Cambridge, UK, 2005).
(6) R.D. McDowall, Quality Assurance J. 9, 196–227 (2005).














