What Is Data Integrity Training?
Spectroscopy
A current hot topic in regulated GXP laboratories is data integrity. Recently, the UK regulator issued an industry guidance on this subject that requires the establishment of a data governance system, part of which is data integrity training. Easy to say, but how do you do it?
A current hot topic in regulated good practice (GXP) laboratories is data integrity. Recently, the UK regulator issued an industry guidance on this subject that requires the establishment of a data governance system, part of which is data integrity training. Easy to say, but how do you do it?
Starting from the beginning, data integrity is a critical component of data, information, and reports submitted to regulatory authorities for licensing applications and also for use in releasing batches of material to the market. If a problem is found with data integrity it casts a shadow over all the work performed by a laboratory. As seen from a warning letter: “The lack of reliability and accuracy of data generated by your firm is a serious CGMP [current good manufacturing practice] deficiency that raises concern for all data generated by your firm” (1).
More noncompliance issues centered on laboratory computerized systems were discussed in this column (2) previously; that column installment focused on technical issues such as user types and access privileges, audit trails turned on, electronic records protection, and lack of procedural controls. In January and March 2015, the UK regulatory body the Medicines and Healthcare products Regulatory Agency (MHRA) issued two versions of a guidance for industry (3,4). In this guidance the MHRA suggested the establishment of a data governance system.
MHRA Data Governance System
The MHRA guidance document defines data governance system as, “the sum total of arrangements to ensure that data, irrespective of the format in which it is generated, is recorded, processed, retained and used to ensure a complete, consistent and accurate record throughout the data lifecycle” (4).
An expansion of the requirements for a data governance system is provided in the definitions section of the MHRA guidance (4):
- Data governance should address data ownership throughout the lifecycle, and consider the design, operation, and monitoring of processes and systems to comply with the principles of data integrity including control over intentional and unintentional changes to information.
- Data governance systems should include staff training in the importance of data integrity principles and the creation of a working environment that encourages an open reporting culture for errors, omissions, and aberrant results.
- Senior management is responsible for the implementation of systems and procedures to minimize the potential risk to data integrity, and for identifying the residual risk, using the principles of the International Conference on Harmonization (ICH) Q9
- Contract givers should perform a similar review as part of their vendor assurance program (4).
OK, great words-let’s explore what they mean.
- No other regulatory agency is requiring organizations to have a data governance system. Regulators require and want data integrity-that is a given-but a data governance system is unique to the MHRA.
- Because of the global data integrity issues being found as a result of data falsification or just poor data management practices, serious consideration needs to be given to a data governance system.
- Within a pharmaceutical quality system (PQS), which is required by European Union Good Manufacturing Practice (EU GMP) Chapter 1 (5) and Annex 11 (6), there are explicit data integrity requirements for both paper and electronic systems as well as requirements in Chapters 4 and 6 for paper, hybrid, and electronic records (7,8). Also, there are implicit Food and Drug Administration (FDA) requirements for data integrity in 21 CFR 211.194(a) (complete data) and 211.68(b) (9).
- A data governance system covers all processes involved in generating data and records during the course of pharmaceutical supply, manufacturing, testing, and release. The controls to be applied to individual records, especially critical ones, are determined by risk management, which will outline the data integrity approaches for each system and process (4).
By analyzing these statements and other portions of the MHRA guidance, we can abstract the main elements of a data governance system. These are summarized below and shown diagrammatically in Figure 1:


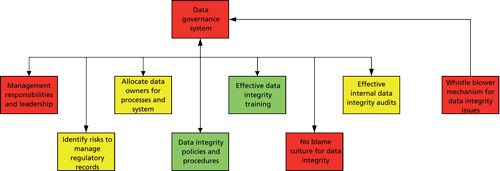
- Ensure senior and laboratory management leadership for data integrity
- Allocate a data owner for manual and computerized data processes
- Risk assessment to identify and manage regulatory records
- Write policies and procedures for data integrity
- Ensure effective data integrity training to ensure the procedures above are followed
- Create a no-blame culture around data integrity-own up to a mistake, don’t cover it up
- Establish a confidential whistle blower avenue for staff to communicate issues to senior management
- Effective internal data integrity audits
An extreme example of a data governance system can be found in the January 2012 Ranbaxy consent decree (10). This established the office of chief data integrity officer reporting to the board with a number of tasks to carry out to resolve the falsification issues that had arisen previously.
Data Integrity Is the Focus
The focus of this column is on the procedures and associated training for data integrity within the framework of either a pharmaceutical quality system or a data governance system. From Figure 1 we can see the elements described below.
Policies, Procedures, and Training
Procedures for ensuring data integrity for all activities (both GMP and non-GMP to avoid dual standards) followed by training in these procedures for all staff is essential (9,11). Data integrity must be included in the regulatory requirement for on-going GMP training to reinforce the message. Part of the policies and procedures is the requirement for risk assessment. This needs to be undertaken to determine the impact and criticality of the records generated by each system to determine the controls. Then, via a gap and plan process, assess the existing controls in place to determine what, if any, additional controls are required to ensure data integrity. The good automated manufacturing practice (GAMP) good practice guide on “Compliant Part 11 Records and Signatures” (12) already has a list of controls to protect electronic records and this could be adapted by organizations to include paper records as well.
Data Ownership
The MHRA guidance requires a data owner (4). Rather than create another role, I would suggest that for computerized systems the existing process owner in the laboratory should also be the data owner. They would be responsible for the integrity of data in their systems.
Regulatory Requirements for GMP Training
The GMP requirement for training under the US regulations is 211.25(a), which states, “Training in current good manufacturing practice shall be conducted by qualified individuals on a continuing basis and with sufficient frequency to assure that employees remain familiar with CGMP requirements applicable to them” (9).
The EU GMP requirement in Chapter 2 (11) is similar.
Industry typically interprets “frequently” as “annually.” However, the regulations are vague: training in CGMP as applicable to an individual’s role. Given the current regulatory interest in data integrity, only the stupid or foolhardy will argue that the subject should not be included in initial or updated GMP training or any quality training. However, the questions arise of what procedures are required or what should the data integrity training consist of? The regulations and regulatory guidance are silent, leaving it to individual companies to draw up their own individual approaches. Perhaps it time to write that awful phrase-let’s think outside of the box.
It is said that plagiarism is the sincerest form of flattery. However, if one is plagiarizing from two or more sources it becomes research. What we need to do is look outside of the pharmaceutical industry and see what others are doing in this area. For instance, let’s consider environmental analysis; my research in this area is discussed in the next section.
Environmental Analysis and Data Integrity
The United States Environmental Protection Agency (US EPA) regulates environmental analysis through its version of good laboratory practice (GLP) regulations (13). In 1990, with increasing computerization it was apparent that there was little in the EPA GLP regulations to control them. Therefore, the EPA initiated a program to ensure the integrity of computer-resident data in laboratories by developing standards for automated laboratory processes. At the start of this program, the EPA commissioned a survey of automated clinical laboratories under the assumption of a high degree of data integrity as they were regulated and inspected by a variety of state authorities and professional organizations. The survey, carried out by consulting firm Booz Allen & Hamilton, highlighted a wide range of controls from very lax to very stringent within the six laboratories surveyed (14).
This survey was an input to the development of the good automated laboratory practice (GALP) guidelines issued by the EPA in draft for comment in 1992 and the final version was released in 1995 (15). Interestingly, the draft version was far more stringent than the released version. GALP has expired now; it has fallen off its perch and now is pining for the fjords; however, the document is still available on the EPA website (16) and reading it provides an interesting compare and contrast with the current GXP approach to data integrity.
Let us move the narrative forward a few years to when a national accreditation program for environmental laboratories was established in 2006 with the formation of The NELAC Institute (TNI) (17). This program was formed from two organizations, one of which was NELAC or the National Environmental Laboratory Accreditation Conference. In 2003, NELAC developed a standard for laboratory accreditation (18) that includes requirements for laboratory data integrity and is the subject of the remainder of this column. This standard is being superseded by a consensus one that is currently under development by TNI (19). However, for our discussion, the 2003 standard is fit for purpose because it contains requirements that pertain to data governance, data integrity, and, specifically, the requirements for data integrity training. For this reason, it will help regulated GXP organizations understand the requirements for data integrity training and they can adapt the approach for their own uses.
NELAC Quality System
The NELAC standard document contains a lot about the conference that established it and the organizational structure of the organization, but our immediate interest is in section 5 dealing with the “Quality System Requirements.” Figure 2 shows the four main data integrity elements within the quality system of the NELAC standard (18):


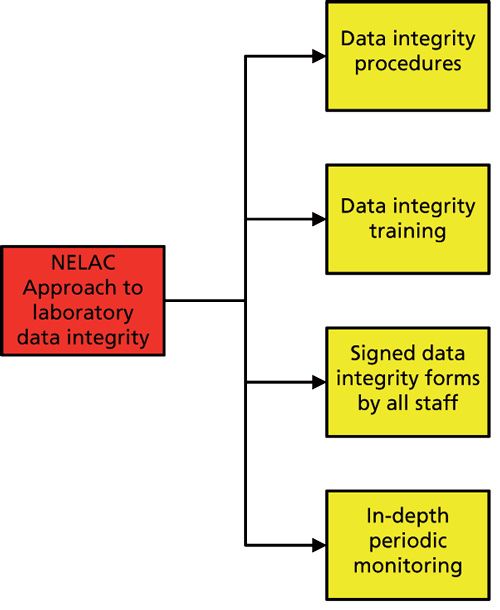
- Data integrity procedures
- Data integrity training
- Signed data integrity forms for all staff
- Periodic in-depth monitoring
Table I presents a précis of the NELAC requirements for data integrity. These sections of the standard take the four elements above and break them down into more detail. The two main elements to consider for an approach to data integrity are that data integrity starts at the top and data integrity reviews are essential. A few key points in these elements are explained below.


Data Integrity Starts at the Top
- Senior management are responsible for the establishment and maintenance of the data integrity procedures.
- Management commitment and support for data integrity is required throughout an organization.
- There must be the ability of staff to raise data integrity issues confidentially for discussion with management.
Data Integrity Reviews Are Essential
- Routine assessment (second person review) of data generated in the laboratory must include checks for data integrity of the data generated.
- In-depth data integrity checks as part of the internal audits conducted by an organization.
- Documented data integrity investigations in cases of suspected breaches of the data integrity procedures with follow-up disciplinary and corrective actions as appropriate.
NELAC Data Integrity Training
Missing from the list above are the detailed requirements for data integrity training, and these are presented in Table II and diagrammed in Figure 3. As you can see, there are very specific requirements for data integrity training for both new and existing employees. The NELAC standard also mentions ethics as well as data integrity training, my interpretation is that the two terms are interchangeable in the context that the right analytical result is supported by the right data records and documentation. Right in the second use of the word in the last sentence encompasses all data, all calculations, and all processed data that have been generated following the right analytical method and the applicable data integrity procedures.





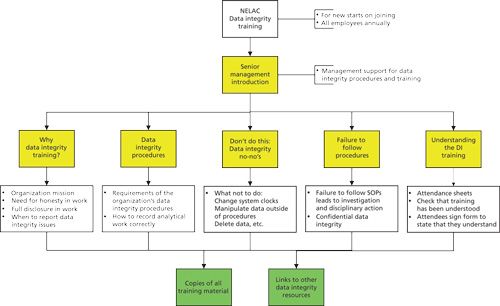
As shown in Figure 3, in my opinion, the training must be introduced by a senior manager because this is the only way to demonstrate that the management team is serious about data integrity. As senior management are responsible for establishing and maintaining data integrity procedures as well as supporting data integrity in the organization, then there should not be a problem for one of them to introduce, at least, any data integrity training session.
In my opinion, the NELAC requirement for understanding the training can only be accomplished by face-to-face training rather than a read and understand or even a computer-based training. My rationale for this is the importance of data integrity training and the need to answer questions interactively that can never be accomplished by a simple read and understand or a computer-based training. The subject is too important to consider any other alternatives.
(Please note that the contents of Tables I and II are my summary of the NELAC standard requirements and are not quoted verbatim. Therefore, if you wish to see the original wording that I have summarized please read the standard itself [18].)
Summary
In this column we have looked at what training integrity could entail for a regulated healthcare laboratory. Starting from the current GMP regulatory requirements for training and the UK’s MHRA data integrity guidance for data governance, we saw a problem with a lack of information on the subject. However, by looking at environmental analysis we discovered the existing NELAC quality system where data integrity is a mandatory requirement. Training in the subject is a specific requirement for all employees when they join and on an annual basis. I have summarized the overall approach that NELAC mandates in its quality system for data integrity and for training in the subject, specifically. Some readers working in healthcare laboratories may view this as a draconian approach for the pharmaceutical industry. However, the genie is out of the pharmaceutical bottle.
Because of either poor data management practices or data falsification in the industry, the regulators are mobilized. The MHRA is the first guidance to be issued by a regulator. I expect guidance or regulations to follow from the FDA, the Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme (PIC/S), and the EU in the short-to-medium term. The pharmaceutical industry needs to respond and not simply by paying lip service on the subject. Management commitment and data integrity procedures with effective employee training are the foundation of this response. Failure is not an option.



References
(1) US Food and Drug Administration, USV Limited, FDA Warning Letter (February 2014).
(2) R.D. McDowall, Spectroscopy29(11), 22–27 (2014).
(3) Medicines and Healthcare products Regulatory Agency, GMP Data Integrity Definitions and Guidance for Industry, Version 1 (MHRA, January 2015).
(4) Medicines and Healthcare products Regulatory Agency, GMP Data Integrity Definitions and Guidance for Industry, Version 2 (MHRA, March 2015).
(5) European Commission Health and Consumers Directorate-General, Eudralex: The Rules Governing Medicinal Products in the European Union, Volume 4: Good Manufacturing Practice, Medicinal Products for Human and Veterinary Use; Chapter 1, Pharmaceutical Quality System (Brussels, Belgium, 2013).
(6) European Commission Health and Consumers Directorate-General, Eudralex: The Rules Governing Medicinal Products in the European Union, Volume 4: Good Manufacturing Practice, Medicinal Products for Human and Veterinary Use; Annex 11, Computerised Systems, (Brussels, Belgium, 2011).
(7) European Commission Health and Consumers Directorate-General, Eudralex: The Rules Governing Medicinal Products in the European Union, Volume 4: Good Manufacturing Practice, Medicinal Products for Human and Veterinary Use; Chapter 4, Documentation (Brussels, Belgium, 2011).
(8) European Commission Health and Consumers Directorate-General, Eudralex: The Rules Governing Medicinal Products in the European Union, Volume 4: Good Manufacturing Practice, Medicinal Products for Human and Veterinary Use; Chapter 6, Quality Control (Brussels, Belgium, 2014).
(9) Code of Federal Regulations (CFR), Part 211, Current Good Manufacturing Practice for Finished Pharmaceutical Products Consent Decree of Permanent Injunction, Ranbaxy Laboratories and Food and Drug Administration, Case 1:12-cv-00250-JFM, United States Court District of Maryland, January 2012.
(10) European Commission Health and Consumers Directorate-General, Eudralex: The Rules Governing Medicinal Products in the European Union, Volume 4: Good Manufacturing Practice, Medicinal Products for Human and Veterinary Use; Chapter 2, Personnel (Principle and clause 2.11) (Brussels, Belgium, 2014).
(11) ISPE, GAMP Good Practice Guide Validation of Laboratory Computerized Systems, Part 11 Records and Signatures (International Society of Pharmaceutical Engineering, Tampa, Florida, 2005).
(12) Code of Federal Regulations (CFR), Part 160, Good Laboratory Practice Standards, Environmental Protection Agency (U.S. Government Printing Office, Washington, DC, 1997).
(13) Automated Laboratory Standards: Evaluation of the Standards and Procedures used in Automated Clinical Laboratories (Contract 68-W9-0037), Booz Allen Hamilton, 1990.
(14) Environmental Protection Agency, Good Automated Laboratory Practice Guidelines (EPA, Washington, D.C., 1995).
(15) Environmental Protection Agency (EPA), expired policies, available at: http://www.epa.gov/irmpoli8/expiredpolicies/2185.pdf.
(16) The NELAC Institute (TNI), available at: www.nelac-institute.org.
(17) National Environmental Laboratory Accreditation Conference, Quality Standard (NELAC, 2003).
(18) TNI information on current standards development, available at: http://www.nelac-institute.org/content/CSDP/standards.php.



R.D. McDowall is the Principal of McDowall Consulting and the director of R.D. McDowall Limited, as well as the editor of the “Questions of Quality” column for LCGC Europe, Spectroscopy’s sister magazine. Direct correspondence to: spectroscopyedit@advanstar.com













