What’s New in the New USP ?
The new version of United States Pharmacopeia general chapter “Analytical Instrument Qualification” became effective August 1, 2017. What does this mean for you?
United States Pharmacopeia general chapter <1058> “Analytical Instrument Qualification” has been updated and became effective August 1, 2017. So, what has changed in the new version?
In the regulated world of good manufacturing practice (GMP) we have regulations that define what should be done, but leave the interpretation to the individual organization on how to do it. However, when we come to the regulated analytical or quality control (QC) laboratory we also have the pharmacopoeias, such as the European Pharmacopoeia (EP), Japanese Pharmacopoeia(JP), and United States Pharmacopeia (USP), to provide further information to help interpret the regulations. These tomes can have monographs for active pharmaceutical ingredients, finished products, and general chapters that provide requirements for how to apply various analytical techniques, such as spectroscopy.
In the Beginning . . .
Of the major pharmacopoeias, only USP has a general chapter on analytical instrument qualification (AIQ) (1). This chapter came about with a 2003 conference organized by the American Association of Pharmaceutical Scientists (AAPS) on analytical instrument validation. The first decision of the conference was that the name was wrong and it should be analytical instrument qualification (AIQ). The conference resulted in a white paper (2) that after review and revision became USP general chapter <1058> on AIQ effective in 2008. This chapter described a data quality triangle, general principles of instrument qualification, and a general risk classification of analytical equipment, instruments, and systems. General chapter <1058> did not specify any operating parameters or acceptance limits because those can be found in the specific general chapters for analytical techniques.
This column is written so that you will understand the changes that come with the new version and the impact that they will have on the way that you qualify and validate instruments and the associated software, respectively.
Why Is Instrument Qualification Important?
The simplest answer to this question is that qualification is important so that you know that the instrument is functioning correctly and that you can trust the results it produces when it is used to analyze samples.
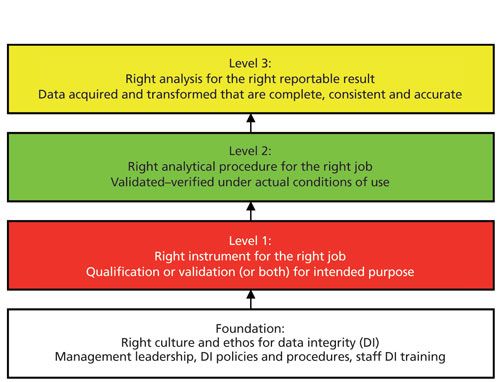
However, there is a more important reason in today’s world of data integrity–integrated instrument qualification and computer validation is an essential component of a data integrity model. The complete four-layer model can be viewed in my recent book, Validation of Chromatography Data Systems (3), but the analytical portion of the model is shown in Figure 1 and is described in more detail in an earlier “Focus on Quality” column (4).
The four layers of the model are
- Foundation: management leadership, policies and procedures, culture, and ethos
- Right instrument and system for the job: instrument qualification and computer validation
- Right analytical method for the job: development and validation of analytical procedures
- Right analysis for the right reportable result: analysis from sampling to reporting the result
The model works from the foundation up with each layer providing input to the next. As shown in Figure 1, AIQ and computerized system validation (CSV) come after the foundation layer, illustrating that if the instrument is not qualified and any software is not validated, the two layers above (method validation and sample analysis) will not be effective and can compromise data integrity. Interestingly, AIQ and CSV are missing from many of the data integrity guidance documents but you can see the importance in the overall data integrity framework of Figure 1.


Figure 1: The four layers of the data integrity model for laboratories within a pharmaceutical quality system. Adapted with permission from reference 4.
Why Is There a New Revision of USP<1058>?
The 2008 version of USP <1058> had several issues that I presented during a session co-organized by Paul Smith at an AAPS conference in 2010 and published in this column (5). There were three main problems with the first version:
- Problem 1: The true role of the supplier was missing. The supplier is responsible for the instrument specification, detailed design, and manufacture of the instrument but this responsibility is not mentioned in <1058>. The reason is that the section on design qualification (DQ) mentions that a user can use the supplier’s specification. However, a user needs to understand the conditions under which the specification was measured and how relevant it is to a laboratory’s use of an instrument.
- Problem 2: Users are responsible for DQ. USP<1058> places great emphasis on the fact that in the design qualification stage is the responsibility of the supplier, “Design qualification (DQ) is most suitably performed by the instrument developer or manufacturer.” This is wrong. Only users can define their instrument needs and must do so to define their intended use of the instrument and comply with GMP regulations (§211.63) (6).
- Problem 3: Poor software validation guidance. The software qualification and validation description in USP <1058> is the poorest part of this general chapter as software is pervasive throughout Group B instruments and Group C systems.
Although the approach to handling embedded software in Group B instruments where the firmware is implicitly or indirectly validated during the instrument qualification is fine, there are omissions. Users need to be aware that both calculations and user defined programs must be verified to comply with GMP requirements in 211.68(b) (6). Note that the qualification of firmware, which is a simple and practical approach, is now inconsistent with Good Automated Manufacturing Practice (GAMP) 5, which has dropped Category 2 (firmware).
Software for Group C systems is the weakest area in the whole chapter <1058>. The responsibility for software validation is dumped on the supplier: “The manufacturer should perform DQ, validate this software, and provide users with a summary of validation. At the user site, holistic qualification, which involves the entire instrument and software system, is more efficient than modular validation of the software alone.”
In the days of data integrity, this approach is completely untenable. The United States Food and Drug Administration (FDA) guidance on software validation (7), quoted by <1058>, was written for medical device software, which is not configured unlike much of the laboratory software used today.
Revision of USP <1058>
To try and rectify some of these issues, the revision process of USP <1058> started in 2012 with the publication of a “Stimulus to the Revision” process article published in Pharmacopeial Forum written Chris Burgess and myself (8). This article proposed two items:
- An integrated approach to analytical instrument qualification and computerized system validation (AIQ-CSV), and
- More granularity for Group B instruments and Group C computerized systems to ensure that calculations and user defined programs were captured in the first group and an appropriate amount of validation was performed by the users for the second group.
We used the feedback from that article to draft a new version of USP <1058> in the summer of 2013, which was circulated to a few individuals in industry and suppliers for review before submission to the USP.
Proposed drafts of the new version were published for public comment in Pharmacopeial Forum in 2015 (9) and 2016 (10) and comments were incorporated in the updated versions. The approved USP <1058> final version was published in January 2017 in the First Supplementto USP 40 (11). The chapter became effective on August 1, 2017. There was an erratum published in February, but the only change was reference of the operational qualification (OQ) testing to the intended use definition (user requirements specification [URS]).
What Has Changed in USP <1058>?
First let us look at the overall scope of changes between the old and new versions of USP <1058> as shown in Table I.


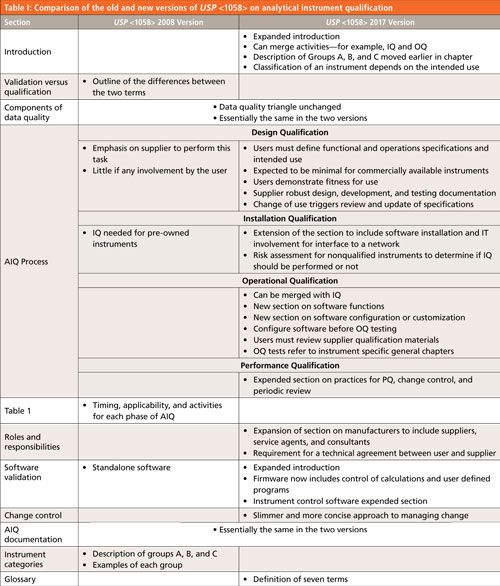
CLICK TABLE TO ENLARGE
Missing in Action
The following items were omitted from the new version of <1058>:
- Differences between qualification and validation. This was omitted because qualification and validation activities are integrated in the new version, so why describe the differences? You need to control the instrument and any software and if you can demonstrate this through the 4Qs process described in the new <1058>, why bother with what the activity is called?
- Table I in the old version of <1058> describes the timing, applicability, and activities of each phase of AIQ and it has been dropped from the new version. Rather than give a fixed and rigid approach to AIQ, as the table did, there is more flexibility in the new version of <1058> and omitting this table reinforces the new approach.
- Standalone software: Because standalone software has nothing to do with AIQ, not surprisingly this section has been omitted from the new version. The GAMP 5 (12) and the accompanying good practice guide for laboratory computerized systems (13) or my chromatography data system (CDS) validation book (3) will be adequate for this task.
- Examples of instruments in the three categories: Because the instrument classification depends on the intended use there is no need to give a long list of instruments or systems in Groups A, B, and C. It is the intended use of the instrument that defines the group and providing a list produces anomalies. For example, in the old version of <1058> a dissolution bath is listed in Group C when it should be in Group B if there is only firmware and the instrument is calibrated for use. To avoid these arguments the list of examples has been dropped from the new version.
Additions and Changes to USP <1058>
Of greater interest to readers will be the changes and additions to the new general chapter, again these can be seen in Table I. Below I will discuss the following three areas that reflect the main changes to the general chapter:
- Roles and responsibilities
- Changes to DQ, installation qualification (IQ), and OQ phases and how this impacts your approaches to AIQ
- Software validation
Roles and Responsibilities
The USP <1058> update to the “Roles and Responsibilities” section makes users ultimately responsible for specifying their needs, ensuring that a selected instrument meets them and that data quality and integrity are maintained (11). The manufacturer section now includes suppliers, service agents, and consultants to reflect the real world of instrument qualification. One new responsibility is for the supplier or manufacturer to develop meaningful specifications for users to compare with their needs. Incumbent on both users and suppliers is the need to understand and state, respectively, the conditions under which specifications are measured to ensure that laboratory requirements can be met. We will discuss this further under the 4Qs model in the next section.
Finally, there is a requirement for a technical agreement between users and suppliers for the support and maintenance of any Group B instrument and Group C system. The agreement may take the form of a contract that both parties need to understand the contents of and the responsibilities of each.
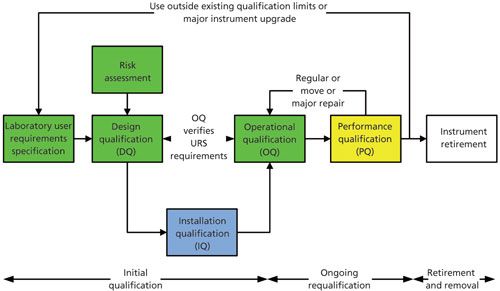
An Updated 4Qs Model
At first sight, the new version of USP <1058> uses the same 4Qs model as the 2008 version. Yes . . . but there are some significant differences. Look at Figure 2, which presents the 4Qs model in the form of a V model rather that a linear flow. This figure was also published in a “Questions of Quality” column authored by Paul Smith and myself (14). However, Figure 2 has now been updated to reflect the changes in the new version of USP <1058>. Look at the green-shaded boxes to see the main changes:


Figure 2: Modified 4Qs model for analytical instrument qualification. Adapted with permission from reference 14.
Design Qualification
Design qualification now has two phases associated with it.
- The first phase is for the users to define the intended use of the instrument in a user requirements specification: Users must define functional and operation specifications and intended use (11). Although the new <1058> notes that this definition is expected to be minimal for commercially available instruments it does not mean slavishly copying supplier specifications-especially if you do not know how any of the parameters have been measured. The output from this process is a URS or your intended use definition.
- The second phase is the qualification of the instrument design. This means that you confirm that the selected instrument meets your design specification or intended use. If looking outside of the analytical laboratory, medical device manufacturers call this process design verification-ensuring that the users’ current requirements are in the system and any omissions are mitigated, where appropriate.
These two sections are where most laboratories get it wrong for reasons such as we know what we want (therefore why bother to document it) or we believed the supplier’s literature. This is where most qualifications fail because there is no specification upon which to base the testing in the OQ phase of the process as shown in Figure 2. Executing an OQ without a corresponding URS or design document is planning to fail any qualification. This is one of the major changes in the new version of USP <1058>.
- Note that when the new <1058> talks about minimal specifications for commercial instruments, it does not include minimal specifications for software used to control them for Group C systems. Here you not only need to consider the control of the instrument, but also the acquisition, transformation, storage, and reporting of data and results that includes how data integrity and quality is assured. This activity is not expected to be minimal-especially in today’s regulatory environment.
- There is also the possibility, for Group B instruments, of merging the laboratory user requirements and the DQ: the decision to purchase being made on the URS (with the proviso that the contents are adequate).
- Risk management is implicit in the <1058> classification of the instrument groups and the subgroups of B and C instruments and systems but more needs to be done in the specification and configuration of the software. For example, access controls, data acquisition, and transformation are key areas for managing data integrity risks.
- What is also shown in Figure 2 is that if there is any change of use during the operation of the instrument or system, it must trigger a review of the current specifications with an update of them, if appropriate.
Installation Qualification
In the new version of <1058>, installation qualification now includes
- The installation of software and the involvement of the IT function to interface an instrument to a network
- The requirement for conducting an IQ for nonqualified instruments is replaced with a requirement to gather available information and conduct a risk assessment to determine if an IQ should be conducted. In many cases, if an instrument has been installed and maintained by a supplier with records of these activities, but has not been formally qualified, the risk assessment may determine that no IQ should be performed, placing increased emphasis on the OQ phase to demonstrate fitness for intended use.
- The introduction to <1058> mentions that activities can be merged-for example, IQ and OQ (11). I would see suppliers taking advantage of this option to have a single IQ and OQ document for ease of working, because both phases are typically conducted by the same individual. The combined protocol must be preapproved by the laboratory and then post-execution reviewed by them. However, the post-execution review needs to be conducted while the engineer is still on site so that any corrections can be done before the individual leaves the site.
Operational Qualification
Operational qualification has also been extended to include
- A new section on software functions and the differences between software configuration and customization for Group C systems.
- It is important to configure software (and document the settings) before an OQ is conducted, otherwise you’ll be repeating some tests. In practice, however, there may be a differentiation of duties because a supplier may only perform a basic qualification of the unconfigured software leaving the laboratory to configure the application and then conduct further verification of the whole system. This point is critical because data integrity gaps are usually closed through configuration of the application. However, unless the application is known or a copy is already installed and configured, it is unlikely that the OQ will be performed on a configured version because the laboratory may not know the process to be automated (for example, hybrid operation, electronic operation, or incorporate spreadsheet calculations into the software).
Software Validation Changes
The major changes to this General Chapter occur in the section on software validation. They are shown diagrammatically in Figure 2. Because the instrument examples have been removed in the new version of <1058> and replaced with the need to determine the group based on intended use, a formal risk assessment now needs to be performed and documented. A risk assessment, based on Figure 3 to classify instruments based on their intended use has been published by Burgess and myself (15) and is based on the updated classification used in the new version of USP <1058> (11). As can be seen from Figure 2, the risk assessment should be conducted at the start of the process in the DQ phase of work because the outcome of the risk assessment can influence the extent of work in the OQ phase.


Figure 3: Software validation and verification options with the new USP <1058>. Adapted with permission from reference 3.
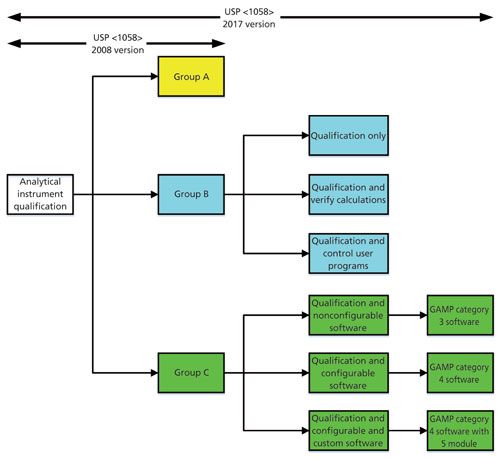
Rather than classify an item as either Group B or Group C, there is now more granularity for both groups with three suboptions in each of these two groups. This increased granularity allows laboratories more flexibility in qualification and validation approaches, but also fills the holes from the first version of <1058>.
Group B instruments now just require either qualification of the instrument and either verification of any embedded calculations if used or specification, build, and test of any user defined programs.
For Group C systems, the new USP <1058> divides software into three types:
- Nonconfigurable software
- Configurable software
- Configurable software with custom additions
As can be seen from Figure 3, these three subtypes can be mapped to GAMP software categories 3, 4, and 4 plus category 5 modules. These changes now align USP <1058> closer to, but not identically with GAMP 5. The main difference is how firmware in Group B instruments is validated-directly with GAMP 5 or indirectly when qualifying the instrument with USP <1058>. Mapping of GAMP 5 software categories to the new USP <1058> groups has been published (16) for those readers who want more information harmonizing <1058> and GAMP approaches. This chapter is much improved and closer in approaches, but not quite there yet!
However, the bottom line is that software validation of Group C systems under the new USP <1058> should be the same as any GxP system following GAMP 5. One item that is not mentioned in the new <1058> is a traceability matrix. For Group B instruments, it will be self-evident that the operating range of a single parameter will be tested in the OQ. However, this process changes with Group C systems, especially because software and networking are involved; a traceability matrix will be mandatory.
Summary
This column has highlighted the main changes in the new version of USP <1058> on analytical instrument qualification that became effective on August 1, 2017. We then discussed three of the main changes: roles and responsibilities, changes to the 4Qs model, and the much-improved approach to software validation for Group C systems.
In general, the USP is moving toward full life cycle processes. When the new version of <1058> became effective in August 2017, it is likely that a new revision cycle will be initiated. If this occurs, a full life cycle will be the centerpiece of this revision.
Acknowledgments
I would like to thank Chris Burgess, Mark Newton, Kevin Roberson, Paul Smith, and Lorrie Schuessler for their helpful review comments in preparing this column.
References
- General Chapter <1058>, “Analytical Instrument Qualification,” in United States Pharmacopeia 35-National Formulary 30 (United States Pharmacopeial Convention, Rockville, Maryland, 2008).
- AAPS White Paper on Analytical Instrument Qualification 2004, (American Association of Pharmaceutical Scientists, Arlington, Virginia).
- R.D. McDowall, Validation of Chromatography Data Systems: Ensuring Data Integrity, Meeting Business and Regulatory Requirements, Second Edition (Royal Society of Chemistry, Cambridge, UK, 2017).
- R.D. McDowall, Spectroscopy31(4), 15–25 (2016).
- R.D. McDowall, Spectroscopy25(11), 24–31 (2010).
- Code of Federal Regulations (CFR), 21 CFR 211, “Current Good Manufacturing Practice for Finished Pharmaceutical Products” (Food and Drug Administration, Sliver Spring, Maryland, 2008).
- US Food and Drug Administration, Guidance for Industry General Principles of Software Validation (FDA, Rockville, Maryland, 2002).
- C. Burgess and R.D. McDowall, Pharmacopeial Forum38(1) (2012).
- “USP <1058> Analytical Instrument Qualification in Process Revision,” Pharmacopeial Forum41(3) (2015).
- “USP <1058> Analytical Instrument Qualification in Process Revision,” Pharmacopeial Forum42(3) (2016).
- General Chapter <1058>, “Analytical Instrument Qualification,” in United States Pharmacopeia 40, 1st Supplement (United Stated Pharmacopeial Convention Rockville, Maryland, 2017).
- ISPE, Good Automated Manufacturing Practice (GAMP) Guide Version 5 (International Society of Pharmaceutical Engineering, Tampa, Florida, 2008).
- ISPE, Good Automated Manufacturing Practice (GAMP) Good Practice Guide: A Risk-Based Approach to GXP Compliant Laboratory Computerized Systems, Second Edition (International Society of Pharmaceutical Engineering, Tampa, Florida, 2012).
- P. Smith and R.D. McDowall, LCGC Europe 28(2), 110–117 (2015).
- C. Burgess and R.D. McDowall, Spectroscopy28(11), 21–26 (2013).
- L. Vuolo-Schuessler et al., Pharmaceutical Engineering34(1), 46–56 (2014).



R.D. McDowall is the Principal of McDowall Consulting and the director of R.D. McDowall Limited, as well as the editor of the “Questions of Quality” column for LCGC Europe, Spectroscopy’s sister magazine. Direct correspondence to: SpectroscopyEdit@UBM.com













