From Heavy Metals Testing to the Measurement of Elemental Impurities in Pharmaceuticals: Over 100 Years in Making the Change
Spectroscopy
A look at the implementation of the new United States Pharmacopeia (USP) chapters and the International Conference for Harmonization (ICH) guidelines for elemental impurities from an historical perspective, providing insight into the changes and considering the challenges and opportunities that lie ahead as the industry embraces the new methodology.
As a follow up to March 2018's "Atomic Perspectives" column, which provided guidance on how to choose the optimum technique for carrying out the measurement of elemental impurities in pharmaceuticals, this month's installment takes a look at the implementation of the new United States Pharmacopeia (USP) Chapters and the International Conference for Harmonization (ICH) guidelines for this method from an historical perspective. This column provides insight into these changes and takes a look at the challenges and opportunities that lay ahead as the industry embraces this new methodology.
The identification and quantification of elemental impurities in pharmaceuticals have been a challenge for many years. In the late 1800s and early 20th century, lead was an issue because it was used in the production of solder, and, absent the good manufacturing practices controls that are in place today, metals from manufacturing and processing equipment posed potential problems, as did mined excipients that could carry with them elements such as arsenic, lead, cadmium, and mercury.
The United States Pharmacopeia (USP) first addressed this issue in 1908, with the publication of what became general chapter <231> - Heavy Metals. This test typically involves the high-temperature digestion of the material of interest followed by sulfide precipitation of the metals of interest. The specification is based on comparison of the color of the resulting precipitate to the color of a lead sulfide precipitate prepared at the level of interest (typically 10 or 20 ppm). There are, of course, many issues with this test. The test generates hydrogen sulfide, which can be highly toxic, and in the USP method, does this through the use of thioacetamide, which has its own safety issues. The test was initially designed to detect lead, mercury, bismuth, arsenic, antimony, tin, cadmium, silver, copper, and molybdenum. Not included are common components of manufacturing equipment such as nickel and vanadium, or common catalysts such as platinum or palladium. In addition, the test is a screening test and is not element specific, and since each precipitate has a different color (and most are lighter than the color of lead sulfide), even its use as a total metal limits test can be questioned. A final issue is that the test is based on visual acuity, rather than being toxicologically based. For a test for elements considered toxic, it is far preferred that it be based not on what one observes, but rather on what levels are of toxicological concern.
The Process for Change
Given all these issues, why did the test remain as an official test in the USP until January 2018, approximately 110 years after its introduction? The primary reason is likely that there was not an industry-wide, globally accepted test to replace it. For all its faults, the methodology of general chapter <231> was well understood, routinely used by the industry, and was harmonized with the European and Japanese pharmacopeias. The winds of change began with the publication in 1995 of a Stimuli to the Revision Process article in the Pharmacopeial Forum by Katherine Blake (1). In this article, she noted that often 50% of the metal of interest was lost in the ash process, and that there was essentially zero recovery for mercury, one of the more toxic and common elements of interest. In 2000, Dr. Taibang Wang made similar observations in an article in the Journal of Pharmaceutical and Biomedical Analysis, where he noted "Although still widely accepted and used in the pharmaceutical industry, these methods based on the intensity of color of sulfide precipitation are non-specific, labor intensive, and more often than hoped, yield low recoveries or no recoveries at all" (2). Then, in 2004, Nancy Lewen and coworkers directly compared the recoveries of 14 elements using USP <231> Method II and inductively coupled plasma-mass spectrometry (ICP-MS). Consistent with the other two articles, this study showed a number of recoveries around 50%, with recoveries of less than 10% for Se, Sn, Sb, and Ru with, as anticipated, no recovery at all for Hg, as exemplified in Figure 1 (3).


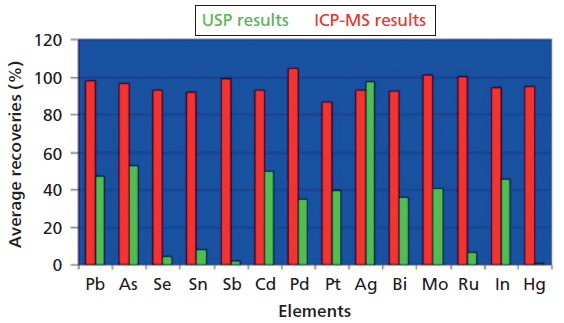
Figure 1: Comparisons between ICP-MS and Method II described in USP Chapter <231> (3).
USP Expert Committees
This work set the stage for the establishment at USP of a series of committees and expert panels, beginning in earnest with the 2005–2010 revision cycle to update the concept of heavy metals testing. The process built on an Institute of Medicine meeting commissioned by USP in 2008, and on a document issued by the European Medicines Agency (EMA), which developed guidelines on the control of residual catalysts in pharmaceuticals with the goal of establishing limits based on toxicological safety assessments of common catalytic elements. This work began as early as 1998 and resulted in a guideline that was officially implemented in 2008 (4).
ICH Involvement
Building on all this, USP published in the Pharmacopeial Forum a proposed general chapter on Inorganic Impurities: Heavy Metals that was the early precursor to general chapter <232> - Elemental Impurities – Limits, and <233> - Elemental Impurities – Procedures (5). This USP initiative led to the desire by the pharmaceutical industry participants to have, as with general chapter <231>, a chapter that was harmonized across the pharmacopeias in the International Conference for Harmonization (ICH) region (at a minimum, the United States, Europe, and Japan). The result of this harmonization initiative was the ICH Q3D expert working group, and the final output was the ICH Q3D guideline on elemental impurities, with the step 4 document posted to the ICH website in December 2014 (6). The guideline was implemented for new drug products by the U.S. Food and Drug Administration (FDA) beginning in July, 2016, and for all drug products, new and existing, in the USP in January 2018, concurrent with omission from the USP of general chapter <231>. General chapter <232> applies only to drug products and not to drug substances or excipients, since without knowledge of issues such as dose and route of administration, establishment of specifications for these materials is not possible.
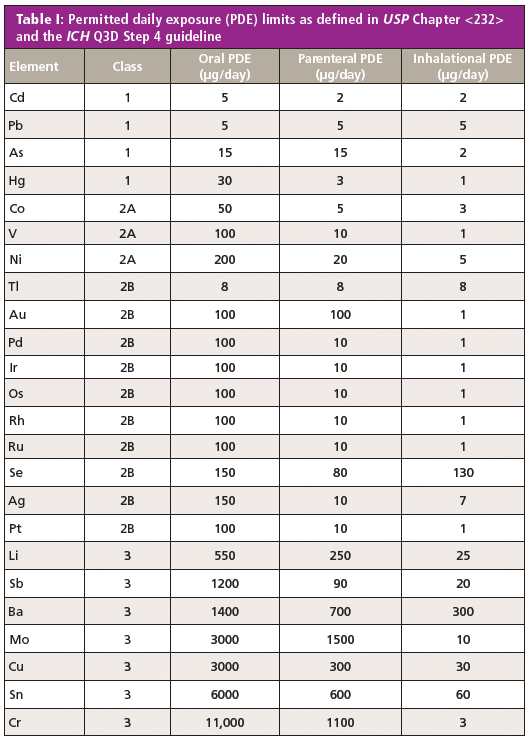
ICH Q3D focused on the establishment of a list of elements of toxicological concern and their limits in oral, parenteral, and inhalational dosage forms as well as a risk-based approach for demonstrating absence of the various metals and compliance with the specification. An extensive body of training materials is available to help practitioners understand the scope of the guideline, the concept of a risk-based approach, and how to think about issues such as the calculations of concentrations in the drug product and extension to other dosage forms (7). Because the entire process is risk based, testing is only one of many ways by which compliance with the standard can be demonstrated. The full list of permitted daily exposure (PDE) elemental limits defined in USP Chapter <232> and the ICH Q3D Step 4 guideline are shown in Table I. Note that the classification is based on their toxicological impact (a combination of toxicity and likelihood of occurrence), with Class 1 and 2A elements considered the most significant.


Testing of drug products and excipients was not discussed in detail by the ICH Q3D working group. Rather, it was left to the individual pharmacopeias to work out their own procedures, with the goal of long-term harmonization of the final results. In the USP, general chapter <233> describes two procedures, inductively coupled plasma–optical emission spectroscopy (ICP-OES) and ICP-MS, along with system suitability and validation requirements for these tests and any alternative tests. It is noted that any test that meets the validation requirements and is otherwise fit for purpose can be used instead of the two proposed tests.
The implementation of the ICH Q3D guideline and the two USP general chapters, and the elimination of USP <231> are major steps forward in the analysis of elemental impurities in pharmaceuticals. There is now broad agreement across the pharmaceutical industry and across many countries about the elements of toxicological concern and the levels at which they should be controlled.
Why Did This Process Take So Long?
Let's get back to the earlier question: Why did it take 110 years to get here? The simple answer is that it took that amount of time and innovation for the right technology to be developed and for the technology to be widely available and accepted by the pharmaceutical industry. For a drug product specification to be accepted as a public standard (as opposed to a private standard between a pharmaceutical company and a regulatory body where the company can perhaps propose a sophisticated test not readily available), any proposed test needs to be well established, well accepted by the industry, reliable, and broadly available, either in house or though contract research organizations.
Until the last several years, these requirements simply hadn't been met. Only a few technologies are capable of meeting the requirements of general chapters <232> and <233>. While ICP-OES was widely available, there are situations, especially for low-dose drugs or parenteral or inhalational drugs, where a number of elements need to be measured or controlled; ICP-MS is the option of choice. Only recently have the software, hardware, price, and availability-either in house or through contract research organizations-been there to make broad applicability across the industry feasible.
In addition, a suite of new technologies, sampling accessories, and data processing tools is making life easier for pharmaceutical companies that are working in a good manufacturing practice (GMP) environment. Some of these are aimed at laboratories that are looking to maximize sample throughput by using productivity enhancement tools to automate the full suite of quality control and spike recovery testing procedures described in Chapter <233>, as well as to ensure that the calculations are done correctly and properly documented, in order to minimize operator interaction, and allow the analyst to focus on other tasks.
Other areas that have been addressed include software tools to provide data in a suitable compliant form for regulatory submission and inspection. For example, any analytical instrument used for measuring pharmaceuticals must also comply with the FDA's 21 CFR Part 11 regulations to ensure the integrity of any electronic records that are generated. The instrument's software control and data handling must therefore incorporate tools to maintain the integrity of the analytical method and subsequent results and provide transparency to data generation, including support for audit trails as well as security features to ensure that alterations cannot be made without a clear explanation of the process. It's therefore very important that the instrument and its software provide the necessary support to meet these kinds of demands in the highly regulated pharmaceutical industry environment.
Risk Assessment
The widespread availability of this technology, and the study and publication of results of the levels of elemental impurities in drug substances and excipients will likely lead to less testing of materials in the long run. Currently, many drug substances and excipients are being tested for the first time. As databases are developed and made public, many excipients and drug substances may be demonstrated to be of sufficiently low concern that they will no longer need to be considered in a risk assessment, and risk assessments may be limited to perhaps a few specific mined excipients, or materials used in very high quantities in a dose form. Thus testing, which is already only one part of an overall risk assessment, may become even less frequent in the future. This development is an example of the technology providing a breakthrough in the assessment of safety, and soon being relegated to a role as a tool for specific purposes and very seldom, if ever, as a tool for routine monitoring of drug products.
What's left to get to the point where the industry feels that the risks of elemental impurities are well understood and controlled and only minimal levels of testing are necessary? A key piece will be accurate measurement of the levels of elemental impurities in drug substances and excipients, and the broad availability of this information. Another key to making this happen is the generation of good data by the people producing data on the various drug substances, excipients, and drug products. The industry needs good data upon which to make decisions, and the generation of such data for elemental impurities is quite difficult. Issues such as sample homogeneity, sample contamination, instrument sensitivity, and instrument artifacts all play a role in the generation of reliable data.
Sample contamination is of particular concern. As discussed above, the elements of most concern (for example, As, Pb, Cd, Hg, Ni, V, and Co) have been selected because they are both potentially toxic and widely found in the laboratory or in nature, whether in glassware, equipment, reagents, or even clothing or accessories worn by analysts or others in the laboratory. Accessories containing metals can be of particular concern. In addition, reagents such as concentrated acids need to be tested to be free of metals to avoid sample contamination. At the trace levels at which these elements are being detected, very low levels of contamination can lead to false positive results, which in turn can result in additional testing and rework.
Similarly, ICP-MS instrumental artifacts can also lead to erroneous results, such as the effect of the 40Ar35Cl polyatomic spectral interference on the measurement of 75As. This effect is of particular concern if chloride is present in the sample matrix or if concentrated hydrochloric acid is used for the digestion of the sample. The chloride ions will combine with the argon from the plasma to form an argon chloride spectral interference that will significantly interfere with the monoisotopic arsenic ion at mass 75. This effect can be minimized by avoiding hydrochloric acid in the digestion procedure, but if this is not possible, or chloride is present in the drug product, an ICP-MS system fitted with a collision–reaction cell is a very important tool to have at your disposal to help prevent the formation of the interference.
In Summary
We have come a long way in the assessment of risk of toxicity from low-level trace elements in pharmaceuticals from the days of sulfide precipitation and comparison to a lead sulfide standard. The new risk assessment strategies and tools-such as closed-vessel digestion to provide analyzable samples with minimal analyte loss and ICP-MS to provide the requisite sensitivity and selectivity-have enabled the industry to quantify the elemental impurity levels in drug substances, excipients, and drug products and reach a point where solid, risk-based assessments can be made with only minimal testing needed in the long term.
This month's column has been adapted from Robert Thomas's new book (8).
References
(1) K. Blake, Pharmacopeial Forum 21(6), 1632–1637 (1995).
(2) T. Wang, J. Pharm. Biomed. Anal. 23, 867–890 (2000).
(3) N. Lewen et al., J. Pharm. Biomed. Anal. 35, 739–752 (2004).
(4) European Medicines Agency, Committee for Medicinal Products for Human Use. 2008. Guideline on the specification limits for residues of metal catalysts or metal reagents. Doc. Ref. EMEA/CHMP/SWP/4446/20007.
(5) General Chapter on Inorganic Impurities: Heavy Metals, PF 34(5), 2008.
(6) ICH website: http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html (Q3D).
(7) ICH website: http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html (Q3D training).
(8) R.J. Thomas, Measuring Elemental Impurities in Pharmaceuticals: A Practical Guide (CRC Press, Boca Raton, Florida, 2018). ISBN13:978-1-138-19796.


Tony DeStefano

Tony DeStefano is a consultant in analytical, compendial, bioanalytical, and quality-related issues. He has worked in the pharmaceutical industry for more than 40 years. As a former Vice President at US Pharmacopeia, he led the initiatives to maintain, update, and redesign the General Chapters, and also served as a member of the ICH Q3D Expert Working Group on metal impurities. He has advanced degrees in Physical Chemistry from Cornell University and is a member of the American Chemical Society (ACS) and past president of the American Association of Pharmaceutical Scientists (AAPS).


Robert Thomas
Robert Thomas is principal of Scientific Solutions, a consulting company that serves the application and writing needs of the trace element user community. He has worked in the field of atomic and mass spectroscopy for more than 40 years and has written over 80 technical publications including a 15-part tutorial series on ICP-MS. He recently completed his fourth textbook entitled Measuring Elemental Impurities in Pharmaceuticals: A Practical Guide. He has an advanced degree in analytical chemistry from the University of Wales, UK, and is also a Fellow of the Royal Society of Chemistry (FRSC) and a Chartered Chemist (CChem). He has served on the ACS Committee on Analytical Reagents for the past 17 years.






Applications of Micro X-Ray Fluorescence Spectroscopy in Food and Agricultural Products
January 25th 2025In recent years, advances in X-ray optics and detectors have enabled the commercialization of laboratory μXRF spectrometers with spot sizes of ~3 to 30 μm that are suitable for routine imaging of element localization, which was previously only available with scanning electron microscopy (SEM-EDS). This new technique opens a variety of new μXRF applications in the food and agricultural sciences, which have the potential to provide researchers with valuable data that can enhance food safety, improve product consistency, and refine our understanding of the mechanisms of elemental uptake and homeostasis in agricultural crops. This month’s column takes a more detailed look at some of those application areas.





