Probing the Structural Effects of Pasteurization and Spray Drying on Soy Protein Isolate in the Presence of Trehalose Using FT-IR-ATR and FT-Raman Spectroscopy
The authors present the results of a study in which FT-IR-ATR and FT-Raman spectrosopies were used to probe the effects of pasteurization and spray drying on the secondary structure of soy protein isolate.



Fourier transform infrared–attenuated total reflectance (FT-IR–ATR) and FT-Raman spectroscopies were used to probe the effects of pasteurization and spray drying on the secondary structure of soy protein isolate (SPI). Deconvolution of the infrared Amide I band was accomplished using a curve-fitting method based upon a simple model and experimentally determined constraints. This method indicated 57% β-sheet and β-turn, 5% α-helix, and 38% random coil for pasteurized, spray-dried SPI. These values are compared with those for "native" SPI, where "native" SPI is estimated using the weight-averaged values for native glycinin and β-conglycinin. From this comparison, we conclude that pasteurized, spray-dried SPI contains less α-helix, more random coil, and similar amounts of β-sheet relative to the "native" SPI. Problems with dispersibility of pasteurized, spray-dried SPI in beverages were attributed to denaturation of native α-helix segments of SPI with subsequent formation of aggregated β-sheets. FT-IR–ATR and FT-Raman results showed that co-drying SPI with trehalose inhibits both the amount of protein denaturation and β-sheet aggregation associated with pasteurization and spray drying. Because denaturation and aggregation are directly correlated to dispersibility (and other bulk properties), controlling these parameters will enable beverage manufacturers to produce products with greater consumer acceptability and improved shelf life.
Soy protein isolate (SPI) is a highly purified form of protein (>90% w/w) obtained from soybeans. It can be incorporated in liquid or dry foods including beverages, nutritional bars, snacks, meats, and bakery goods. In most cases, it is convenient to produce SPI as a dry material. Producing dry SPI on an industrial scale typically involves the following processes: aqueous extraction of defatted soy flour or flakes, precipitation, separation, pasteurization, and spray drying. Unfortunately, pasteurization and spray drying subject food proteins to thermal stress. Depending upon residence time and temperature, proteins can undergo irreversible denaturation, and upon reconstitution, proteins may aggregate, reducing dispersibility in beverages. To minimize this undesirable effect, it is helpful to understand how the protein structure is altered by pasteurization and spray drying. In the first part of this study, we used Fourier transform infrared (FT-IR) and FT-Raman spectroscopies to reveal changes in the secondary structure of SPI associated with pasteurization and spray drying.
It is well known that trehalose stabilizes protein structure during thermal treatment (1). Our group is interested in identifying and understanding ways in which trehalose can be used to improve functionality of thermally processed food proteins. In the latter part of this study, FT-IR and FT-Raman spectroscopies were used to probe the ability of trehalose to inhibit denaturation and aggregation of soy proteins during pasteurization and spray drying. The results will be used to help facilitate improvements in product functionality when SPI is incorporated in beverages.
Experimental
Materials used included SPI (Prolisse, Cargill, Inc., Wayzata, Minnesota) and trehalose (Ascend, Cargill, Inc.). Aqueous SPI samples were spray dried with trehalose. Trehalose was added after an ultrafiltration step and just before a pasteurization step (110 °C for 12 s) that precedes spray drying (240 °C in, 80 °C out). The lyophilized SPI was collected as an aqueous sample before the pasteurization step and freeze dried with a Labconco Freezone 6-L freeze-dry system (Labconco Corp., Kansas City, Missouri).
FT-IR
FT-IR spectra of protein powders were acquired at 2-cm–1 resolution with a Varian FTS 7000 spectrometer (Varian, Randolph, Massachusetts) equipped with a narrow-band mercury cadmium telluride (MCT) detector. Each interferogram was collected with 256 scans, apodized with a medium Beer–Norton function, and Fourier transformed with no zero filling before interpretation (several levels of zero filling are used for presentation). Six replicate measurements were made using six aliquots to minimize effects from heterogeneity and ensure reproducibility. The final spectrum was obtained by averaging the six replicates. Sampling was accomplished with a temperature-controlled diamond anvil attenuated total reflectance (ATR) accessory (SPECAC, Woodstock, Georgia) and a sapphire anvil. For each measurement, excess sample (~100 mg) was placed on the ATR crystal with the anvil well above the sample while the hinged piece was secured. The anvil was then lowered straight down onto the "mound" of powdered sample. Good reproducibility was achieved with this procedure. Both the spectrometer and the ATR accessory were purged with dry air.
FT-Raman
FT-Raman spectra were acquired at 8-cm–1 resolution with an FT-Raman accessory coupled to the Varian FTS 7000 spectrometer, using a 1064-nm, 2-W Nd:YAG laser and a liquid nitrogen–cooled Ge detector. Each interferogram was collected with at least 256 scans, apodized with a medium Beer–Norton function, and Fourier transformed with no zero filling before interpretation (several levels of zero filling are used for presentation). The boiled-to-dryness SPI was sampled on a glass slide. All other samples were placed in standard 5-mm diameter nuclear magnetic resonance (NMR) tubes (approximately 4.2 mm i.d.) and, to avoid photodecomposition and heterogeneity errors, were rotated at ~100 rpm during measurement using a customized sample spinner built in-house. For each sample, six separate spectra were acquired at different positions along the tube height (while spinning) to further ensure representative bulk sampling. The final spectrum was obtained by averaging the six replicates.
Spectral manipulations: Spectral manipulations were performed using GRAMS/32 (Galactic Industries, Salem, New Hampshire). The second derivative spectra were calculated using a two-degree, 15-point Savitzky–Golay smoothing algorithm.
Deconvolutions: Deconvolutions were performed with MatLab (Natick, Massachusetts) using a curve-fitting methodology that was developed in-house. While it is beyond the scope of this article to cover all of the analysis methods in detail, each of the steps involved in producing the Amide I curve fits will be summarized here, with emphasis placed on those aspects of the fitting procedure that are most crucial for obtaining reliable secondary structure results.
First, we subtract residual water vapor and carbon dioxide peaks from each of the sample spectra by fitting reference spectra of these constituents using an entropy minimization algorithm (2). Then, we remove artifacts due to ATR sampling effects (such as frequency-dependent depth of penetration, sample density, and baseline curvature) from these spectra by applying a weighted extended multiplicative scatter correction (WEMSC) (3,4). Specifically, frequency regions known to consist primarily of absorbance from carbohydrate and protein constituents are weighted to zero so that they do not bias the WEMSC calculation. (Note that SPI is >90% protein.)
The actual curve-fitting step employs the conjugate gradient implementation from the Optimization Toolbox in Matlab, except that we modify it to include a linear fit step of the peak amplitude parameters within each nonlinear fitting iteration (5,6). This interim step saves time by reducing the number of nonlinear parameters that need to be iteratively fit. For example, applying a model consisting of four Gaussian–Lorentzian mixture bands in the Amide I region to a set of 50 spectra implies initially that 800 parameters must be found, because the four peaks in each of the 50 spectra consist of four parameters: an amplitude, a width (fwhm), a position, and a Gaussian–Lorentzian relative fraction (with 1 being full Gaussian and 0 being full Lorentzian). However, for the model to be realistic, the same four peaks should fit all 50 spectra equally well; only the peak amplitudes should change between spectra consisting of perturbations of a single protein type. This means that the model can be simplified to only 12 nonlinear parameters (the width, position, and Gaussian–Lorentzian ratio for each of the four peaks), and 200 linear parameters (the four peak amplitudes for each of the spectra). Redefining the curve fit model in this way improves the fidelity of the resultant fits by taking advantage of inherent correlations among the spectra and by reducing the chance of "overfitting" any one spectrum in the data set.
The curve-fit model includes peaks to account for overlap in the Amide I region from Amide II, amino acid side-chain and lipid carbonyls, but we do not explicitly interpret these. Also, the model includes terms for fitting a quadratic-shaped baseline to account for any residual ATR artifacts. The curve-fit calculation is initialized with peak positions taken from features in the average of the inverted second derivatives of the processed spectra. Initial peak widths are set at 30 cm–1 , and Gaussian–Lorentzian fractions are set at 0.5. These starting values are based upon our own experience and consensus in the literature. We use three diagnostics to determine the quality of the fit. First, we apply the parsimony principle: we include only as many peaks as are required to yield normally distributed fit residuals (that is, the reduced χ2 statistic is ~1). Second, we verify that there are no extra or misplaced peaks by checking that the inverted second derivatives of the fit spectra match those of the sample spectra. Finally, we ensure that our calculated secondary structure percentages are in approximate agreement with independent sources, such as X-ray diffraction studies.
Molecular models: X-ray diffraction data were obtained as PDB files from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank. Molecular protein models were produced using Pymol (DeLabno Scientific LLC, San Carlos, California).
Results and Discussion
Glycinin and β-conglycinin account for about 70% of the protein in soy protein (7,8). Molecular models of glycinin and β-conglycinin were created using X-ray diffraction data for the native state proteins and are presented in Figure 1 (9,10). We use these models to reveal important information about the nature of the molecules and to help establish constraints for FT-IR–ATR Amide I curve-fitting models (discussed later). The α-helices are presented in blue, the β-sheets in green, and the random coil conformations in purple. For both proteins, note the stable β-sheet network that forms the core of the protein, and the significant portion of the α-helix conformation that is located outside of the core. With this information, we might expect a loss of α-helix with the onset of thermal denaturation, while the β-sheets' content remains relatively constant. This is indeed the behavior observed and reported for pure glycinin (11,12).


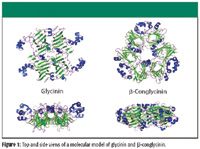
Figure 1
The Amide I region (near 1650 cm–1 ) of both infrared and Raman spectra is sensitive to the secondary structure of proteins and can be used to monitor changes in protein conformation (13,14). The ν(OH) region (near 3300 cm–1 ) of the infrared is very sensitive to changes in water and can be used to monitor changes in water content. FT-IR–ATR spectra of lyophilized and pasteurized, spray-dried SPI are presented in Figures 2a and 2b, respectively. Lyophilization is a milder process than spray drying and produces fewer structural changes in the soy proteins. Because the lyophilized sample experiences a lesser degree of thermally induced denaturation and aggregation when drying and the sample was not subjected to the pasteurization process, the lyophilized SPI should retain more native structure than the pasteurized, spray-dried SPI. We therefore use it as a pseudo control. Comparison of the spectra shown in Figures 2a and 2b reveals differences in intermolecular β-sheet (discussed later).



Figure 2
Pasteurized, spray-dried SPI that was mixed with water and heated to dryness on the ATR crystal at 90 °C is presented in Figure 2d. Pasteurized, spray-dried SPI that has been repeatedly subjected to a similar treatment at 125 °C is presented in Figure 2c. Comparison of the spectra shown in Figures 2b and 2d reveals that the SPI treated at 90 °C contains more β-sheet and less water than the SPI (ν[OH] region not shown). Spectrum 2C reveals that the SPI treated repeatedly at 125 °C contains large numbers of random coil conformations and thus is mostly unfolded.
FT-Raman spectra of lyophilized and pasteurized, spray-dried SPI are presented in Figures 3a and 3b, respectively. An FT-Raman spectrum of pasteurized, spray-dried SPI that has been mixed with water and heated to dryness at ~100 °C on a glass slide is presented in Figure 3c. Comparison of the spectra shown in Figures 3a and 3b reveals differences associated with a broad feature near 1685 cm–1 . The feature likely originates from a band that is too broad and has a frequency that is too high to be associated with β-sheet (or β-turns). The shape and position of the feature are consistent with the shoulder of a random coil band, which appear as very broad Amide I bands in the Raman spectra (15). Furthermore, the feature is more prominent in the spectrum of pasteurized, spray-dried SPI, which is the more denatured sample. The appearance of this band is reasonable because previous studies suggest that random coil increases as α-helix decreases in the initial stages of thermally induced denaturation (11,12). We therefore assign the origin of the feature to the broad random coil band. Comparison of FT-Raman spectra shown in Figures 3b and 3c also reveals that after additional heat treatment, the pasteurized, spray-dried SPI contained more β-sheet, thus showing that the changes observed in the FT-IR–ATR spectra are not due simply to decreasing water content. We were unable to obtain an FT-Raman spectrum analogous to the FT-IR–ATR spectrum 2C, because our larger scale preparation procedure eventually browns the SPI before reaching this state, and the respective FT-Raman spectra become compromised by fluorescence.


Figure 3
Using a model and a deconvolution procedure, it is possible to quantify changes in α-helix, random coil, and β-sheet (16). In this work, we used a curve-fitting approach based upon a simple model with experimentally determined constraints (see experimental section for details). The deconvolved FT-IR Amide I spectrum of pasteurized and spray dried SPI is presented in Figure 4.



Figure 4
Although the Amide I band profile is rich in structural information, numerous challenges and complications exist for extracting accurate quantitative results (17). Of special note is the strong absorption from condensed phase water at ~1640 cm–1 , which requires spectral subtraction. A second technique is often required to minimize ambiguities. In our work, we use FT-Raman spectroscopy to provide qualitative confirmation. Water is a relatively weak Raman scatterer and therefore produces significantly less interference in the Raman spectrum. Furthermore, the individual secondary structure bands appear differently in Raman spectra, which is very useful for confirmation.
The Amide I contour was obtained by subtracting spectral contributions due to the condensed phase water contribution (~1640 cm–1 ) and the large Amide II contour at 1550 cm–1 . Deconvolution was accomplished by curve fitting with a constrained model that contains only four bands. The deconvolved bands are listed and identified in the legend to the left. Integrated areas indicated 57% β-sheet and β-turn, 5% α-helix, and 38% random coil. These values are compared with those for "native" SPI presented in Table I, where "native" SPI is estimated using the weight-averaged values for native glycinin and β-conglycinin. From this comparison, we conclude that pasteurized, spray-dried SPI contains less α-helix, more random coil, and similar amounts of β-sheet to the "native" SPI. Because the amount of β-sheet remains relatively constant, we further conclude that pasteurization and spray drying have little effect on the stable β-sheet core common to soy proteins. Because the native soy proteins are globular and most of the α-helices are located near the surface of the native protein (Figure 1), loss of α-helix should increase surface hydrophobicity, which might induce aggregation.



Table I: Secondary structure of soy proteins
Soy proteins aggregate during thermal processing in a way that can affect product functionality negatively. Previous studies have shown that part of the aggregation process involves the formation of intermolecular β-sheets (11,12). It is possible to study this type of protein aggregation with FT-IR because the intramolecular β-sheet bands appear at higher frequencies than the intermolecular β-sheet bands (18–20). Differences between lyophilized and pasteurized/spray-dried SPI are observed readily in the second derivative spectra that are presented in Figure 5. In Figure 5, note the larger intensity near 1620 cm–1 for the pasteurized, spray-dried SPI, indicating a larger concentration of intermolecular β-sheet in the spray-dried sample. The spectra have been smoothed to emphasize these features and to deemphasize artifacts associated with small water vapor bands. Changes observed in the 1670-cm–1 band of the second derivative FT-Raman spectra are consistent with increasing total β-sheet due to spray drying (spectra not shown). We now expand our claims concerning thermal denaturation. Specifically, we attribute the lower dispersibility of pasteurized, spray-dried SPI in beverages to denaturation of native α-helix segments of SPI with subsequent formation of aggregated or intermolecular β-sheet formation.


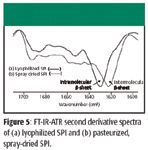
Figure 5
FT-IR spectroscopy was then used to monitor the number of intermolecular β-sheets formed during pasteurization and spray drying as a function of trehalose concentration. To obtain the necessary samples, increasing levels of trehalose were added to SPI before the pasteurization process, which immediately precedes the spray-drying process. Two separate trials were performed at a soy protein processing plant, and quantitative FT-IR–ATR results were obtained by modifying the curve-fitting approach described earlier to include separate bands for intra- and intermolecular β-sheet (aggregated β-sheet) (18). The results showed that trehalose (6–15% w/w powder) reduced β-sheet aggregation up to 30% relative to a control. Plots of %reduction in aggregated β-sheet in pasteurized, spray-dried SPI versus trehalose concentration for two separate trials are presented in Figure 6. From these plots, we conclude that trehalose inhibits the formation of intermolecular β-sheets, and the effect is concentration-dependent.


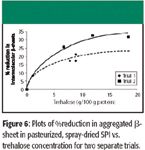
Figure 6
The FT-Raman Amide I spectra associated with Trial 1 are presented in Figure 7, along with the respective concentrations of trehalose present during spray drying. (Note that trehalose does not interfere with the Amide I region of proteins.) A strong, broad feature is observed in the 0% trehalose spectrum near 1685 cm–1 . This is the same feature observed in the spectrum shown in Figure 3b that we assigned previously to random coil. Because the magnitude of this feature decreases systematically with increasing trehalose concentration, we conclude that trehalose inhibits denaturation and the effect is concentration-dependent.


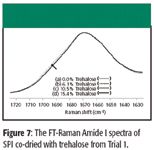
Figure 7
Conclusions
Deconvolution of the FT-IR–ATR spectrum of pasteurized, spray-dried SPI indicated 57% β-sheet and β-turn, 5% α-helix, and 38% random coil. Values for "native" SPI were estimated using the weight averaged values for native glycinin and β-conglycinin and compared with the values for pasteurized, spray-dried SPI. From this comparison, we conclude that the pasteurized, spray-dried SPI contains less α-helix, more random coil, and similar amounts of β-sheet to the "native" SPI. Pasteurization and spray drying reduces dispersibility of SPI in beverages. This problem is attributed to denaturation of native α-helix segments of SPI with subsequent formation of aggregated β-sheets. FT-IR–ATR and FT-Raman results showed that both denaturation and β-sheet aggregation were reduced when SPI was pasteurized and spray dried in the presence of trehalose. Because protein denaturation and aggregation reduce dispersibility of SPI in beverages, controlling these parameters will enable beverage manufacturers to produce products with greater consumer acceptability.
Acknowledgments
The authors would like to thank Mike Porter for providing the lyophilized sample and Jeff Evans for providing the SPI samples that were co-dried with trehalose. The authors also would like to thank Jodi Engleson, Stefan Baier, and Janice Johnson for their support and valuable discussions and Cargill for funding this work.
References
(1) A. Patist and H. Zoerb, Colloids Surf., B 40, 107–113 (2005).
(2) L. Chen and M. Garland, Appl. Spectrosc. 56, 1422–1428 (2002).
(3) N.B. Gallagher, T.A. Blake, and P.L. Gassman, J. Chemom. 19, 271–281 (2005).
(4) A. Kohler, C. Kirschner, A. Oust, and H. Martens, Appl. Spectrosc. 59, 707–716 (2005).
(5) W.H. Lawton and E.A. Sylvestre, Technometrics 13(3), 461–467 (1971).
(6) M. Maeder and A.D. Zuberbuhler, Anal. Chem. 62, 2220–2224 (1990).
(7) R.A. Badley, D. Atkinson, H. Hauser, D. Oldani, J.P. Green, and J.M. Stubbs, Biochim. Biophys. Acta 412, 214–228 (1975).
(8) V.H. Thanh and K. Shibasaki, J. Agric. Food Chem. 26, 692–695 (1978).
(9) M. Adachi, J. Kanamori, T. Masuda, K. Yagasaki, K. Kitamura, B. Mikami, and S. Utsumi, Proc. Natl. Acad. Sci. U.S.A. 100, 7395–7400 (2003).
(10) N. Maruyama, M. Adachi, K. Takahashi, K. Yagasaki, M. Kohno, Y. Takenaka, E. Okuda, S. Nakagawa, B. Mikami, and S. Utsumi, Eur. J. Biochem. 268, 3595–3604 (2001).
(11) P. Robert, C. Mangavel, and D. Renard, Appl. Spectrosc. 55, 781–787 (2001).
(12) E.N.C. Mills, N.A. Marigheto, N. Wellner, S.A. Fairhurst, J.A. Jenkins, R. Mann, and P.S. Belton, Biochim. Biophys. Acta 1648, 105–114 (2003).
(13) S. Krimm and J. Bandekar, Adv. Protein Chem. 38, 181–364 (1986).
(14) R.W. Williams, Methods Enzymol. 130, 311–331 (1986).
(15) H.G.M. Edwards and E.A. Carter, in Infrared and Raman Spectroscopy of Biological Materials, H. Gremlich and B. Yan, Eds. (Marcel Dekker, New York, 2000), pp. 421–476.
(16) D.M. Byler and H. Susi, Biopolymers 25, 469–487 (1986).
(17) M. Jackson and H.H. Mantsch, Crit. Rev. Biochem. Mol. Biol. 30, 95–120 (1995).
(18) A.C. Dong, S.J. Prestrelski, S.D. Allison, and J.F. Carpenter, J. Pharma. Sci. 84, 415–424 (1995).
(19) A.H. Clark and C.D. Tuffnell, Int. J. Pept. Protein Res. 16, 339–351 (1980).
(20) A.H. Clark, D.H.P. Saunderson, and A. Suggett, Int. J. Pept. Protein Res. 17, 353–364 (1981).
Douglas L. Elmore, Sean A. Smith, Carrie A. Lendon, and Allen R. Muroski are with Cargill Global Food Technology Group, Wayzata, Minnesota.















