USP <1058> and the GAMP Guide on Laboratory Computerized Systems – Is Integration Possible?
Here's what needs to be done to harmonize these two documents.



The United States Pharmacopeia general chapter on analytical instrument qualification (USP <1058>) and the ISPE's Good Automated Manufacturing Practice (GAMP) Good Practice Guide on laboratory computerized systems are the two main sources of guidance for qualifying analytical instruments and validating computerized systems used in the laboratory. This column explains the discrepancies between the two documents as well as changes now being made to both in an attempt to enable an integrated approach to qualification and validation of laboratory instruments and systems.
There are many sources of advice on computerized system validation and analytical instrument qualification for the laboratory, including regulatory agencies, such as the United States Food and Drug Administration (FDA) (1,2); regulatory associations such as the Pharmaceutical Inspection Convention/Co-operation Scheme (PIC/S) (3,4); the Official Medicines Control Laboratories (OMCL) in Europe (5); and pharmacopeias such as the United States Pharmacopeia (USP) (6). Information also can be obtained from scientific societies or associations such as the American Association of Pharmaceutical Scientists (AAPS) (7), the Parenteral Drug Association (PDA) (8), the Drug Information Association (DIA) (9), and the International Society of Pharmaceutical Engineering (ISPE) (10). All of these organizations have published guidance on instrument qualification or computer validation either for a general regulated audience or specifically for a regulated laboratory.
There are two main sources, however, of regulatory guidance and advice for qualification of analytical instruments and validation of computerized systems used in the laboratory. The first is USP general chapter <1058> on analytical instrument qualification (AIQ) (6), which was derived from an AAPS meeting on analytical instrument validation held in 2003. One decision that came from that conference was that the terminology being used at the time was incorrect, because the conference name itself should have referred to analytical instrument qualification. The white paper published by AAPS in 2004 (7) was the major input to USP <1058>, which became effective in 2008.
The second source for guidance in a regulated laboratory comes from ISPE's Good Automated Manufacturing Practice (GAMP) Guide, which is seen by many as a standard for computerized system validation. After the publication of version 4 of this guide in 2001 (11), ISPE published several "good practice guides" (GPGs) on specific topics that were intended to take the principles of the version 4 guide and tailor them for a particular subject or focus area. The GAMP Good Practice Guide on the Validation of Laboratory Computerized Systems is one such guide that was published in 2005 (12).
The major problem with analytical instruments that are used in a regulated laboratory is their great variety, complexity, and variations in intended use. Furthermore, the software associated with an instrument can vary from firmware in basic instruments to servers and workstations for multiuser networked data systems.
Writing guidance for the qualification of this wide range of instrumentation and software is not easy, as qualification needs also depend on the intended use of the instrument or system. However, as we have both maintained for a number of years, only an integrated approach to instrument qualification and computer validation — focusing on the key elements that must be controlled in a single combined process — is efficient and effective (13–16). An integrated approach is not only beneficial from a regulatory and auditable context, but it also is cost effective for the business. This approach is in contrast to conducting an initial qualification of the instrument and a separate validation of the software, which is inefficient and may duplicate some work. Furthermore, because of organizational structures, instrument qualification may be carried out by the vendor and considerable time may elapse before the computer validation is conducted and the system is released into operational use.
It would be highly advantageous if the regulations and guidance could all say similar things and avoid duplicate tasks. However, with the way the current versions of USP <1058> (6) and the GAMP laboratory systems GPG (12) are written, this is not possible, as we will illustrate now.
Critique of the GAMP GPG Laboratory Guide
In 2006, comments were made this column on the disconnection of the first edition of the GAMP GPG for validation of laboratory computerized systems from the rest of the regulated organization (13). The version of the GPG at that time had, from our perspective, the following issues:
- The guide had an unnecessarily complex classification of laboratory computerized systems (13) that did not match the software-based classification in the main GAMP guide (17,18).
- According to the GAMP GPG, everything required validation; there was no consideration of instrument qualification.
- There was no linkage with USP <1058> on AIQ.
- There was no reference to the seminal instrument qualification papers, such as the discussion of modular and holistic qualification approaches by Furman and colleagues (19).
To be fair, the GAMP GPG embraced a simplified life cycle model that was a great advance compared to the traditional V model shown in GAMP 4. In 2008, GAMP 5 was released (10) and was an improvement to the previous version of the guide (11). The new GAMP 5 was more risk-based and introduced several life cycle models depending on the software category. However, the major problem with this new version of the GAMP guide was the removal of category 2 (firmware) from the categorization of software (17,18). While understandable from a software system perspective, it is in direct conflict with USP <1058>, in which group B instruments are firmware-based (6).
Critique of USP <1058>
Two earlier "Focus on Quality" columns commented on USP <1058> (13,16). The classification of analytical equipment into one of three groups (A, B, and C) is a simple risk assessment, which is a good approach, but conflicts with the more complex GAMP laboratory GPG. Some of the other problems of <1058> are
- It makes the vendor, rather than the user, responsible for design qualification and defining the intended purpose for a specific laboratory. This is wrong. Only users can be responsible for defining their requirements and selecting the instrument or system that is appropriate to meet their scientific, regulatory, and business needs. The role of a vendor's specification is to sell instruments.
- The true role of the vendor is missing from the data quality triangle in <1058> (16).
- There are subcategories within group B (instruments) and group C (systems) that are not covered in the current version of <1058>. This can lead to noncompliance with 21 CFR 211.68(b) with regard to checking calculations embedded in some group B instruments (16).
- There is no control or guidance in group B instruments for users to program routines.
- There is poor software validation guidance for group C systems that could result in regulatory observations for a laboratory.
- The reference to the FDA's guidance General Principles of Software Validation (1) is inappropriate because the FDA guidance does not consider the software configuration that is common with laboratory computerized systems.
Readers should note that all USP analytical general chapters will be undergoing revision between now and 2015, with updates being published in Pharmacopeial Forum; this revision will be combined with efforts to harmonize with chapter with both the Japanese and European pharmacopeias. New general chapters will be published in pairs: The legal requirements will be in chapters numbered between <1> and <999> and the corresponding best practice will be in a general chapter numbered between <1000> and <1999>.
AIQ and Computerized System Validation: Where Are We Now?
With the current versions of USP <1058> and the GAMP GPG on laboratory computerized systems, if we ask the question, "Is integration possible?" the answer is a resounding no. Specifically, there is no effective and efficient link between USP <1058>, and the GAMP 5, or indeed the GAMP laboratory GPG. So, where do we go from here?
The next steps will take place on two fronts: first, a stimulus to the revision process paper for the update of USP <1058> (20); and second, the drafting of the second edition of the GAMP GPG on the validation of laboratory computerized systems (21), both of which are planned for publication in the first quarter of 2012.
USP <1058> Stimulus to the Revision Process
During the AAPS annual meeting in November 2010, we suggested to the USP that we write a stimulus to the revision process paper on <1058>. Our proposal was accepted and we began working on a draft of the paper, scheduled for publication in Pharmacopeial Forum in the January–February 2012 issue (20). The main aspects of our paper are described below.
Software Is Important in Analytical Instrument Qualification
Two key points are necessary for effective and efficient AIQ. The first is defining the intended purpose of an item of laboratory equipment. The second is identifying the software used in that equipment. Typically, this software is either firmware inside an instrument or on a separate PC running a software application for controlling the instrument, as well as acquiring, interpreting, reporting, and storing the data. Neither of these software elements is adequately covered in the current version of <1058> (16).
Mapping USP <1058> to GAMP 5 Software Categories
One of the first considerations for revising <1058> should be to close the gap in the approaches of GAMP 5 and <1058> to reach a unified approach to qualification and validation, which is shown in Figure 1. This figure shows our mapping of the current GAMP software categories against <1058> groups A, B, and C. Our contention is that there are subcategories within groups B and C that are not covered by the current version of <1058> but that should be there to ensure comprehensive regulatory guidance (16). It is important to realize that USP <1058> is driven by an instrument or hardware approach (classification into Groups A, B, and C). In contrast, the GAMP approach is software driven. When developing laboratory guidance, we have to consider both sides of the equation: hardware and software.



Figure 1: Mapping USP and GAMP software categories.
Dropping GAMP software category 2 requires category 3 to accommodate items ranging from simple analytical instruments with firmware to laboratory computerized systems with nonconfigurable commercial software. Potentially, we would require validating all group B instruments under GAMP rather than qualify them under <1058>. Because group A items do not contain software, there is no comparable mapping possible with GAMP 5, but we have included this group in Figure 1 for completeness. We also have added GAMP software category 5 under category 4 with it offset to the right in Figure 1 to show that with some category 4 systems it is possible to write user-defined macros.
Comprehensive Risk Assessment Process
The basic risk assessment model in <1058> is the classification of all laboratory items into one of the groups (A, B, or C) based on a definition of intended use. This approach is generally sound, because apparatus (group A), instruments (B), and systems (C) are easily classified. However, there is a weakness in that the level of granularity offered is insufficient to classify the variety of instruments (B) and systems (C) used in combination with software in the laboratory today. Figure 1 illustrates this point by depicting three subtypes within group B instruments (that is, firmware, firmware with calculations, and firmware with the ability for users to define programs).
Therefore, our basic proposal in the stimulus to the revision process paper (20) is to provide better means of
- unambiguously differentiating between apparatus (group A) and instruments (group B) based on functionality
- linking the software elements with the various types of instrument (group B) and systems (group C), because current instrumentation is more complex that the simplistic use of groups A, B, and C. This will identify subgroups within groups B and C.
- identifying items used in a regulated laboratory that are not GXP relevant, to exclude them from the qualification and validation process.


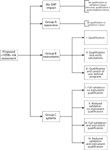
Figure 2: Classification of laboratory items under the proposed risk assessment.
We see this risk assessment as essential for determining the proper business and regulatory extent of qualification and validation for a specific instrument or system with a defined intended use. It also is a necessary requirement for complying with US good manufacturing practice (GMP) regulations, specifically 21 CFR 21.68(b), which requires that calculations be checked if the instrument or system has calculations upon which the user relies (22). This requirement is not mentioned in the current version of <1058>.
The risk assessment we propose is based on asking up to 16 closed questions (with only yes or no answers) that can classify an item in one of the four groups listed below and shown diagrammatically in Figure 2:
1. No GXP impact of the instrument or system
2. Group A (apparatus) — no formal qualification impact, only observation
3. Group B (instruments)
- Instrument only: qualification required (subcategory I)
- Instrument with software: qualification required and calculations verified (sub- category II)
- Instrument with software: qualification required plus control of user-defined programs (subcategory III)
4. Group C (systems)
- Full validation required but without instrument qualification (subcategory I)
- Reduced validation required but without instrument qualification (subcategory II)
- Full validation and instrument qualification required (subcategory III)
- Reduced validation and instrument qualification required (subcategory IV).
Terminology Is Important
You will notice that we talk in terms of apparatus,instruments, and systems for groups A, B, and C, respectively. This is deliberate and is based on the current definitions in <1058>, and also more accurately reflects the items found in these three groups rather than using the all-encompassing term of analytical instruments. We also recommend that the use of the ambiguous term equipment be discontinued in the current context.
4Qs Model Is Replaced by Risk-Based Qualification and Validation
The 4Qs model of instrument qualification is confusing because there are two 4Qs models, which we discuss in the stimulus to the revision process paper: one for instruments, outlined in <1058>; and the second for computerized system validation (CSV), outlined in PDA Technical Report 32 (8). Also, the FDA does not use the terms installation qualification (IQ), operational qualification (OQ), or performance qualification(PQ) in the General Principles of Software Validation (1), as they explain in section 3.1.3 of that document:
While IQ/OQ/PQ terminology has served its purpose well and is one of many legitimate ways to organize software validation tasks at the user site, this terminology may not be well understood among many software professionals, and it is not used elsewhere in this document.
Qualification terminology is also not well understood in the analytical laboratory because readers have to be aware of the context in which a specific term (qualification or validation) is used and although we use the same terms (IQ, OQ, and PQ) they mean different things depending on whether we are talking about qualification or validation (20).
In contrast, both GAMP 5 (10) and the second edition of the laboratory GPG (21) use the term verification, which was adopted from the American Society for Testing and Materials (ASTM) Standard E2500 (23), which includes both the terms qualification and validation as well as the evergreen phrase "fit for intended use" throughout. ASTM E2500 defines verification as a systematic approach to verify that manufacturing systems, acting singly or in combination, are fit for intended use, have been properly installed, and are operating correctly. This is an umbrella term that encompasses all types of approaches to ensure that systems are fit for use in qualification, commissioning, and qualification, verification, system validation, or others (23).
This definition can be compared to the one in ANSI–IEEE standard 610.1990 (24), which defines verification as:
1) The process of evaluating a system or component to determine whether the products of a given development phase satisfy the conditions imposed at the start of that phase; or (2) Formal proof of program correctness
This Institute of Electrical and Electronics Engineers (IEEE) standard has been adopted as an American National Standards Institute (ANSI) standard. Therefore, use of the term is mandatory for all US government departments including the FDA. If we focus only on the first IEEE definition, this can be considered a subset of the ASTM definition of verification as follows: In software engineering, which is the context of IEEE 610, the deliverable or product of a lifecycle phase, say a functional specification, is verified against the input to it (for example, user requirements specification) to ensure that all requirements have been developed into software functions. This is a degree of rigor that is missing in many laboratory validation projects.
GAMP Lab Systems Guide:Second Edition
Since the release of version 5 of the GAMP guide (10), the 2005 version of the laboratory GPG has been out of step with the risk-based approach taken by the former publication. The GAMP forum made a decision to revise the document and publish a second edition of the GPG (21). A team led by Lorrie Schuessler of GlaxoSmithKline (GSK, King of Prussia, Pennsylvania), started the revision process of the GPG.
Scope of the GPG Second Edition
The remit of the GPG team was to revise, not reinvent, the document. One of the key areas was to align the second edition of the GPG with the terms and principles of GAMP 5. In doing this, there was a move from discrete laboratory computerized system categories to a risk-based approach, within which there was an increased emphasis on leveraging assessments and other services provided by instrument suppliers. The team also was tasked with providing ideas for efficiency in validation activities and harmonize with USP <1058>, which was omitted from the first edition of the GPG.
A draft of the proposed GPG was issued for public comment in March 2011 and those comments were incorporated into the revision process. When the GPG team learned of our planned update to USP <1058> they proposed a collaboration to align and integrate the two approaches. We were happy to agree. We worked closely and openly with a core team, including Lorrie Schuessler, Mark Newton, and Paul Smith, to help draft, review, and revise appendixes to integrate as much as possible the GAMP GPG with our proposed update of <1058> (20,21).
Changes in the Second Edition of the Laboratory Systems GPG
A major change to the laboratory systems GPG will be the removal of the categories of laboratory computerized systems. Depending on your perspective they were either loved (what?!) or hated (we're in this camp). In practice, however, both the wide range of instruments and systems as well as the great number of business processes supported made use of categories problematic. The same item could be in several different categories depending on how it was used in a particular laboratory — for example, a sonic bath used for dissolving solutions, for delivering quantitative sonic energy or temperature, or in a robot system. Thus, the wide ranges of use made single categories misleading and failed to effectively use the resources needed for validation. So, now the categories of laboratory computerized systems have been replaced with the relative terms simple, medium, and complex.
The second edition of the GPG is nearly twice the size of the first edition, and the majority of the new content is contained in the appendixes (21). The first edition had only three appendixes, whereas the second edition has 12 appendixes to describe items in more detail. Furthermore, where a topic has been covered in sufficient detail in the main GAMP guide, the reader is referred to it.
New Appendixes
The appendixes of the second edition GPG are listed below:
1. Comparison of USP <1058> and GAMP GPG
2. Categories of Software
3. System Description
4. Application of EU Annex 11 to Computerized Lab Systems
5. Data Integrity
6. Definition of Electronic Records and Raw Data
7. Activities for Simple Systems
8. Activities for Medium Systems
9. Networked Chromatography Data System with Automated HPLC Dissolution
10. Instrument Interfacing Systems including LIMS and Electronic Notebooks
11. Robotics Systems
12. Supplier Documentation and Services
From our point of view, Appendix 1 is the most important because it brings together the two approaches in a single discussion. A good inclusion in the GPG are discussions on the latest regulatory requirements: Appendixes 4 and 6 address the impact of the new EU GMP regulations of Annex 11 (25) and Chapter 4 (26), respectively. The increased emphasis by the regulatory agencies on data integrity also has been addressed, in Appendix 5, which helps laboratories meet the challenge of data integrity in an electronic environment. Validation activities for simple, medium, and complex systems are discussed in four of the appendixes. Finally, there also is a discussion of supplier documentation and services and how to leverage and use them.
AIQ and CSV. Where Will We Be in the First Quarter of 2012?
The title of this column asked if integration of USP <1058> and the GAMP GPG for validation of laboratory computerized systems was possible. With the current versions of the two documents, this is not possible, because of the divergent approaches explained earlier.
However, the first quarter of 2012 brings the promise of integration, because both two publications will be updated at that time. Our stimulus to the revision process paper for USP <1058> will be published in Pharmacopeial Forum and will detail the risk assessment and the subdivision of Groups 1, 2, and 3 (20). The second edition of the GAMP GPG for the validation of laboratory computerized systems also will be published (21). In it, the categories will be eliminated and replaced with the GAMP software categories. Both documents have common elements and approaches, because the teams have collaborated to achieve this.
So, back to the question posed in the title: Is integration possible between <1058> and the GAMP GPG? Yes, it is, and with the updates of these two documents, we are moving toward that ideal. However, life is not perfect, at least not yet. GAMP software category 2 needs to be reinstated for full alignment with <1058> Group B instruments and to allow more explicit flexibility in the laboratory computerized systems GPG. Qualification of laboratory instrumentation is not a term that is recognized by GAMP because they have decided to use verification instead, yet ISPE provides guidance documents on facility commissioning and qualification (27) — so where is the problem? The revision of USP <1058> also uses the term validation, which is avoided in the GPG. However, these differences are easily surmountable with intelligent interpretation and implementation of an integrated approach to AIQ and CSV in your analytical laboratory.
In the future, we hope that we will have USP <1058> providing the regulatory overview of analytical instrument qualification and linking to the relevant requirement chapters of USP that contain the specific instrument parameters to qualify. The GAMP laboratory GPG could then provide guidance on how to achieve this as well as the validation of the software elements (from a single embedded calculation to the whole application or system) — a unified and integrated approach.
If this occurs, then the pharmaceutical industry can meet the existing approach that ISO 17025 (28) states in section 5.5.2:
Equipment and its software used for testing, calibration and sampling shall be capable of achieving the accuracy required...
This implies an integrated approach to ensure that the analytical instrument and the associated software work, as specified for the intended purpose. Nothing more and nothing less.
Acknowledgments
The authors would like to thank the following parties for their contribution to developing the stimulus to the revision process, the second edition of the GAMP Laboratory GPG, and review of this column:
Horatio Pappa, USP
Members of the GAMP GPG for Validation of Laboratory Computerized Systems were Lorrie Schuessler (GlaxoSmithKline), Mark Newton (Eli Lilly & Co.), Paul Smith (Agilent Technologies), Carol Lee (JRF America), Christopher H. White (Eisai Inc.), Craig Johnson (Amgen Inc.), David Dube (Aveo Pharmaceuticals Inc.), Judy Samardelis (Qiagen), Karen Evans and Kiet Luong (GlaxoSmithKline), Markus Zeitz (Novartis Pharma AG), Peter Brandstetter (Acondis) Rachel Adler (Janssen Pharmaceutical), and Shelly Gutt (Covance Inc.).
Mark Newton, Paul Smith, Lorrie Schuessler, and Horatio Pappa for providing comments on the draft of this column.
Chris Burgess has more than 36 years of experience in the pharmaceutical industry, primarily with Glaxo in quality assurance and analytical R&D. He is a qualified person under EU GMP and a member of the United States Pharmacopoeia's Council of Experts 2010–2015. He also is a visiting professor of the University of Strathclyde's School of Pharmacy and Biomedical Sciences in Glasgow, Scotland.



Chris Burgess
R.D. McDowall is the principal of McDowall Consulting and director of R.D. McDowall Limited, and the editor of the "Questions of Quality" column for LCGC Europe, Spectroscopy 's sister magazine. Direct correspondence to: spectroscopyedit@advanstar.com


R.D. McDowall
References
(1) Guidance for Industry, General Principles of Software Validation, FDA (2002).
(2) Guidance for Industry, Computerized Systems in Clinical Investigations, FDA (2007).
(3) Pharmaceutical Inspection Convention and Co-operation Scheme (PIC/S), PIC/S PI-011 Computerized Systems in GXP Environments (2004).
(4) http://www.edqm.eu/en/General-European-OMCL-Network-46.html. A document for HPLC qualification was updated in 2011: http://www.edqm.eu/medias/fichiers/UPDATED_Annex_1_Qualification_of_HPLC_Equipment.pdf.
(5) Pharmaceutical Inspection Convention and Co-operation Scheme (PIC/S), Recommendations on Validation Master Plan, Installation and Operational Qualification, Non-Sterile Process Validation and Cleaning Validation, PI-006 (2001).
(6) United States Pharmacopoeia (USP) <1058> Analytical Instrument Qualification.
(7) American Association of Pharmaceutical Scientists (AAPS) Analytical Instrument Qualification white paper (2004).
(8) Validation of Computer Related Systems, Technical Report 18, Parenteral Drug Association (PDA) (1994).
(9) Computerized Systems used in Non-Clinical Safety Assessment — Current Concepts in Validation and Compliance, Drug Information Association (2008).
(10) Good Automated Manufacturing Practice (GAMP) Guideline Version 4, ISPE (2001).
(11) Good Automated Manufacturing Practice (GAMP) Guideline Version 5, ISPE (2008).
(12) GAMP Good Practice Guide on the Validation of Laboratory Computerized Systems, First Edition, ISPE (2005).
(13) R.D. McDowall, Spectroscopy 21(4), 14–30 (2006).
(14) R.D. McDowall, Spectroscopy 21(11), 18–23 (2006).
(15) R.D. McDowall, Spectroscopy 21(11), 90–95 (2006).
(16) R.D. McDowall, Spectroscopy 25(11), 24–29 (2010).
(17) R.D. McDowall, Spectroscopy 24(6), 22–31 (2009).
(18) R.D. McDowall, Spectroscopy 25(4), 22–31 (2010).
(19) W. Furman, R. Tetzlaff, and T. Layloff, JOAC Int.77, 1314–1317 (1994).
(20) C. Burgess and R.D. McDowall, Pharmaceutical Forum, scheduled for Jan-Feb issue in press 2012.
(21) GAMP Good Practice Guide on the Validation of Laboratory Computerised Systems, Second Edition, ISPE, in press, scheduled for publication Q1 2012.
(22) US GMP 21 CFR 211.68(b).
(23) ASTM Standard 2500, Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment, American Society for Testing and Materials (2007).
(24) IEEE Standard 610.1990 and American National Standard, Glossary of Software Engineering Terminology, IEEE Piscataway (1990).
(25) EU GMP Annex 11 on Computerized Systems (2011)
(26) EU GMP Chapter 4 Documentation (2011).
(27) ISPE Good Practice Guide: Applied Risk Management for Commissioning and Qualification, ISPE (2011).
(28) ISO 17025, General Requirements for the Competence of Testing and Calibration Laboratories, ISO, Geneva (2005).











