Analysis of Lignin and Cellulose in Biological Energy Sources by Raman Microscopy
Cellulosic feedstocks from biological harvests (such as timber, prairie grass, and corn stover) or industrial–urban waste have been proposed as a source for the production of energy in the form of fermentation-produced ethanol biofuel.



With the coming shortages in fuel, the issues of high carbon dioxide emissions, and the problems in waste disposal, society is now searching for alternative, sustainable energy sources. Biological materials are being targeted as attractive feedstocks. They are available in industrial and urban waste streams, in lumber and prairie grasses, and in agricultural products including agricultural waste. The major constituents of plant biomass from these sources are polysaccharides and lignin. To capture high-energy products from these materials, useful chemical forms have to be identified and isolated.


Fran Adar
Ethanol, which is the most common biofuel, has been produced effectively by fermentation from glucose for many years. In particular, corn-derived starch (a glucose polymer) and sucrose (the glucose-fructose dimer derived from sugar cane) have been used as feedstocks. However, it is of questionable sensibility to use agricultural capacity in a world where adequate food resources are becoming scarcer. For this reason, alternate sources of glucose are being investigated.
Starch, the α1,4-polymer of glucose is the primary storage form of gluocse, while cellulose, which is the β1,4-polymer, is the key structural component in plants. Cellulose can provide glucose by freeing the monomer from the cellulose polymer via digestion by fungal cellulase enzymes. But before the cellulose can be digested efficiently, it has to be liberated from lignin and hemicelluloses, the two other polymers that are biologically cosynthesized and serve as essential constituents of the biological matrix. Chemical physical processes that separate and purify the cellulose are being improved to increase their efficiency. Also, the separated components can be further catalytically reformed into hydrocarbons, including aromatic species.
Raman microscopy has a major role to play in monitoring the separation of the cellulose from the lignin and hemicelluloses. Both lignin and hemicellulose are also key biological polymers. Lignin is a branched polymer of three phenols (coumaryl, coniferyl, and sinapyl alcohols). Coumaryl, the basic unit, is a 1,4-substituted aromatic ring with an OH group on one side of the ring, and a –CH=CH2OH on the other. Coniferyl has one methoxy group on one side of the OH, whereas sinapyl has two methoxy groups on either side of the OH. Because of the 1,4 substitution on the ring, the Raman spectrum will not show the usual intense band at 1000 cm-1, but the aromatic signature at 1600 cm-1 will be present. Also present will be the signature of the >C=C< bond near 1650 cm-1. And, in addition, lignin contains acetyl groups, with the carbonyl signature (>C=O) near 1735 cm-1.
Hemicelluloses are a family of cell-wall polysaccharide polymers of molecular weight much lower than that of cellulose. All of the backbones are β1,4-polymers of monosaccharides, usually xylose, mannose and arabinose. Varying degrees of short branches occur with most hemicelluloses.
Hemicellulose and lignin are removed or penetrated by mechanical action to make the cellulose accessible for digestion by the fungal cellulases. However, it is believed that there is a further phenomenon that can affect the accessibility to digestion, and that is the crystallinity of the cellulose. It might be somewhat surprising that a biological polymer can be crystalline, but people who have studied cellulose have documented four crystalline forms and the Raman spectra are different for the different forms. Usually it is cellulose I that occurs naturally. Note that cellulose with large capabilities for hydrogen bonding can be tightly aggregated and quite compact. Based upon more than 30 years of experience using Raman and NMR spectroscopy, as well as X-ray diffraction, to study the structure of cellulose, one of us (Prof. Atalla) has found that the exposure of cellulose to cellulase digestion can be made more effective by chemical–physical methods of preparation of the cellulose, which avoid compaction and crystallization. Raman microscopy also has the potential to distinguish the amorphous and various crystalline forms by monitoring the widths of crystalline-sensitive bands.
It is important to note that Raman microscopy is unique in that it can provide structural information on the constituents without altering or breaking down the structure to isolate individual constituents. Furthermore, because the spectra are dominated by the skeletal vibrational modes of the constituents, the spectra are very sensitive to changes in states of aggregation.
Measurement Protocol
The basis for extracting chemical information from Raman spectra is to follow bands that are characteristic of particular species. However, when there is molecular orientation in the samples being examined, bands can change in relative intensity because of orientation differences rather than chemical differences. In the case of cellulose, the relationship between the molecular axis and the growth axis is quite variable from species to species. So you could either try to randomize your measurements totally or control the orientation–polarization behavior.
For the measurements that will be discussed here, we studied wood chips of young aspen. We tried dispersing fibers on a microscope stage, but found that the dispersal was not randomized, probably because of the effect of the direction of throw on the final chip orientations. So in this study, we will show the results of measurements of individual fibers selected so that their orientation was either parallel or perpendicular to the left–right motion of the microscope stage.
Over the years, we have developed our own protocol for such measurements. While H (horizontal) and V (vertical) are meaningful ways to describe the polarization directions of linearly polarized light, they do not make any sense at the microscope stage because everything is H there. So we have used directions of the map to define the polarization directions for a microscope sample. Light can be polarized EW (east–west) or NS (north–south) at the sample. For most microscope systems, EW will be H and NS will be V before and after the microscope, but there are cases in which this might not be the case (depending how the light is reflected). In addition, it is important to note that any dispersive Raman system that uses a diffraction grating to separate the Raman light into its various components will be inherently dichroic; that is, its sensitivity to the two polarization directions will not be the same. And the value for the dichroism can vary over an order of magnitude depending upon the grating being used and the region of the spectrum being studied. What all this means is that the best way to start such a study would include a total understanding of the behavior of the instrument. But in fact, if conditions are controlled, and all measurements are done in the same manner, useful information can be extracted without systematic artifacts.
Another issue is to confirm the legitimacy of a given measurement. Multiple measurements recorded under identical conditions have been made on fibers extracted from each sample. That is, a series of spectra was recorded from fibers oriented EW, and then a series oriented NS was recorded. For the most part, the differences within a set were far smaller than those between sets. Figure 1 shows such a series. These spectra were recorded with the 532-nm line of a frequency-doubled Nd:YAG laser. (Yes, a green wavelength did produce good spectra!) Before plotting, they were corrected for the instrument response function (filter, grating, and so forth) and then baseline subtracted. (This instrument utilized dielectric edge filters for reflection of the laser line to the sample, and then its suppression in transmission. These filters exhibit ripples in their transmission as a function of wavelength; this behavior was measured and factored out.)


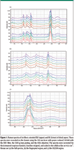
Figure 1
The sample used for these spectra were dried aspen chips without any chemical treatment. The spectrum below 1500 cm-1 is dominated by bands from cellulose. Lignin has three bands of interest: 1600 cm-1 is assigned to the aromatic group, ~1650 cm-1 to the >C=C< bond, and 1730 cm-1 to acetyl groups that are known to be present in lignin. In cellulose, it is known that the ratio of the bands at 1095 and 1122 cm-1 reflect the orientation of cellulose relative to the electric vector (polarization) of the laser. When the electric vector has a component along the cellulose axis, the band at 1095 cm-1 is intensified. So the protocol for the display was to set the intensity of the band at 1122 cm-1 the same for all spectra. The largest variation within a set is in the CH region of the NS-oriented fibers: the fourth spectrum from the bottom of the figure shows unusually high intensity in the saturated CH region of the spectrum (<3000 cm-1). While the other spectra match more closely the published spectrum of cellulose in the CH region, this one does not, which we infer to mean that there was more hemicellulose in the region probed by the laser beam. This is also consistent with the fact that hemicelluloses contain a large amount of five-carbon sugars. Because the pendant CH2 in xylose has its CH2 scissors band near 1450–1460 cm-1, the subtle low frequency shift of this band near 1465 cm-1 indicates a higher content of five-carbon sugars. We do not associate this increase with lignin because lignin is dominated by unsaturated species whose CH stretches are all >3000 cm-1.
Description of samples
To extract cellulose from timber fibers, they are usually first treated with acid chlorite at elevated temperatures (>70 °C). This treatment oxidizes the lignin and releases it into the liquid phase. For the fibers studied here, this treatment was performed at room temperature in order to avoid compaction (crystallization) of the cellulose, which would be expected to occur at elevated temperatures. Further treatment with sodium hydroxide is known to release the hemicelluloses and any residual lignin. We also examined fibers that had been further treated with glycerol at elevated temperatures, which is know to compact the cellulose. The goal was to correlate the Raman measurements with what is known about the chemistry of the fibers. Multiple measurements of the Raman spectra of oriented fibers were compared as shown above and summed in order to compare the behavior of "average" spectrum of each sample type.
Sample measurements
Figure 2 is a summary of the measurements. The disappearance of the lignin bands (between 1575 and 1750 cm-1) is correlated with a known decrease in lignin content. In the CH region, there is a systematic decrease in a band at 2937 cm-1. This is the same band that appeared anomalously high in one of the untreated fiber spectra in Figure 1. Because we know that hemicellulose also decreases with chemical treatments, it makes sense to assign this band to that of hemicellulose. In addition, the aromatic CH at 3067 cm-1 also is seen to disappear. Whether these spectra can answer the question about cellulose compaction will probably require better signal to noise, especially in the region below 600 cm-1.


Figure 2
Summary and the Future
These measurements have shown that by controlling the polarization conditions and the orientation of timber fibers, it is possible to record spectra that are representative of the chemical state of the fibers. The ultimate goal, of course, is to extract quantitative information on the concentration of the components. Using band assignments, we have shown that the spectra are consistent with what is known chemically. What needs to be done now is to take this preliminary work and make numerous systematic measurements to determine the necessary statistics. There are many multivariate analysis algorithms that have been developed for this type of concentration prediction. Because "pure" materials clearly are not available, the best algorithms will probably be unsupervised.
Raj Atalla is with Cellulose Sciences International, Madison, WI.
Fran Adar is the Worldwide Raman Applications Manager for Horiba Jobin Yvon (Edison, NJ). She can be reached by email at fran.adar@horiba.com
References
(1) R.H. Atalla et al., in ACS Symposium Series No 48, Cellulose Chemistry and Technology, J.C. Arthur, Jr., Ed. (ACS, 1977), Ch. 3.
(2) R.H. Atalla et al., Macromolecules 13, 1717–1719 (1980).
(3) R.H. Atalla, in Comprehensive Natural Products Chemistry, Vol. 3, M. Pinto, Ed. (Elsevier, NY, 1999), Ch. 16.












A Proposal for the Origin of the Near-Ubiquitous Fluorescence in Raman Spectra
February 14th 2025In this column, I describe what I believe may be the origin of this fluorescence emission and support my conjecture with some measurements of polycyclic aromatic hydrocarbons (PAHs). Understanding the origin of these interfering backgrounds may enable you to design experiments with less interference, avoid the laser illuminations that make things worse, or both.

Leveraging Electrochemical Impedance Spectroscopy for Lithium-Ion Battery Temperature Prediction
January 17th 2025Researchers have developed a non-invasive, highly accurate method using electrochemical impedance spectroscopy (EIS) to predict the temperature of lithium-ion batteries in real time.


Agilent Presents Awards to 3 Professors for Lithium-Ion Battery Research
January 15th 2025The Solutions Innovation Research awards were presented to Professor Anders Bentien of Aarhus University, Professor Walter Gössler of the University of Graz, and Professor Gregory Offer of Imperial College London.

