A Brief Review of Recent Advances in Isomeric N- and O-Glycomics
Special Issues
Glycan isomer expressions have not been well studied, due to inefficient separation and structural identification techniques. Fortunately, with the development of novel separation techniques and liquid chromatography–mass spectrometry (LC–MS) based glycan isomer identification strategies, new efforts have been made to investigate the glycan isomers in various diseases. Here, we review the recent advances of several isomeric separation techniques for both N- and O-linked glycans.
Glycosylation accounts for the highest abundance of post-translational modifications (PTMs), since more than 50% of mammalian proteins are glycosylated. Recently, glycosylation has attracted much interest due to its versatile biological functions, including cell signaling, cell-cell interaction, cell adhesion, and protein stabilization. Aberrant glycosylation has been demonstrated to be associated with the development of diseases. Thus, many glycomics studies have been performed to investigate glycan expression alterations in biological processes. Furthermore, glycan isomers have also been reported to be involved in many diseases, a fact that demands more in-depth study of isomeric glycomics. However, most research efforts to date have focused on glycan profiles. Glycan isomer expressions have not been well studied, due to inefficient separation and structural identification techniques. Fortunately, with the development of novel separation techniques and liquid chromatography–mass spectrometry (LC–MS) based glycan isomer identification strategies, new efforts have been made to investigate the glycan isomers in various diseases. In this paper, we review the recent advances of several isomeric separation techniques of both N- and O-glycans, along with recent clinical glycan isomer biomarker discoveries, to provide an overview of isomeric glycomics for the benefit of further studies.
Protein glycosylation, as the most abundant post-translational modification in mammalian cells (1–3), mediates many essential biological processes, including cell–cell communication (4,5), cell adhesion (6,7), human immunity (8,9), gene expression (10,11), and protein structural stability (12). The importance of glycosylation is further emphasized by the fact that more than 50% of mammalian cells are glycosylated (13). Recently, aberrant glycosylation has attracted much interest, due to its association with many diseases and disorders (14,15), including inflammation (16,17), Alzheimer's disease (18,19), diabetes (20,21), Parkinson's disease (22), and tumorigenesis (23–26). Although the important role played by glycosylation in bioprocesses has been considered, glycomics remains a significant challenge, due to the microheterogeneity of glycans.
There are two major types of glycans: N-linked glycans (N-glycans), which attach to the asparagine with an amino acid motif of asparagine-X-threonine/serine (NXT/S), where X can be any amino acid except proline; and O-linked glycans (O-glycans), which attach to serine or threonine without a particular consensus (27). Commonly, human glycans are composed of eight monosaccharides, including glucose (Glc), galactose (Gal), mannose (Man), N-acetylglucosamine (GlcNAc), N-acetylgalactosamine (GalNAc), N-acetylneuraminic acid (NeuAc or sialic acid), fucose (Fuc), and glucuronic acid (GlcA) (21,28). Although the glycan building units are limited, glycan structures are complex, because the biosynthesis of glycans is a template-free process that involves many glycosylation enzymes (29). This biosynthesis mechanism results in glycans with different monosaccharide linkages and positions, forming multiple isomers from the same composition.
The presence of enormous numbers of glycan isomers has complicated glycomic analysis due to (i) higher demand for efficient separation techniques; (ii) a lack of effective structural identification methods; and (iii) low abundance of some isomers. However, the biological functions of glycan isomers should not be ignored; many recent studies have demonstrated the relationship between diseases and glycan isoforms (30,31). Alley and Novotny (30) demonstrated that the increase of α2,6 sialylation linkages was significant in cancer progression. Mechref and coworkers observed the significant alterations of glycan isomers in breast cancer cell lines (32) and hepatocellular carcinoma (33). These findings stress the importance of glycan isomer characterization and the possible functions of glycan isomers involved in bioprocesses, beyond glycan profiling. Although the necessity of isomeric glycomics has been realized, the progress of glycan isomer study is hampered due to the need for efficient separation and structural characterization techniques.
Fortunately, isomeric glycomics has developed rapidly alongside improved separation and identification technologies during the last decade (3). The use of mass spectrometry (MS) significantly facilitates glycomic analysis, with its high resolution, high sensitivity, and sufficiency of structural information (34,35). Novel derivatization strategies provide better accuracy and ionization efficiency in glycan identification and quantitation (36). Effective separation techniques such as capillary electrophoresis (CE), high-performance liquid chromatography (HPLC), and ion mobility coupled with MS (IMS) have also prompted the development of glycomics. However, compared to glycan profiling, isomeric glycan analysis is still limited, due to the relative difficulty of glycan isomer separation and characterization. The fact that no glycan isomer has been successfully identified as a biomarker in clinical diagnosis and prognosis denotes that more glycan isomeric studies are necessary, to better understand the biological roles of glycan isomers.
Figure 1 depicts the typical protocol of isomeric glycomics. The principal aim of this review is to discuss recent advances in isomeric glycan studies regarding various separation, derivatization, identification, and quantitation techniques for better isomeric glycan characterization. Efforts to discover glycan isomer biomarkers will also be discussed, in order to provide additional information about further glycan isomeric studies.


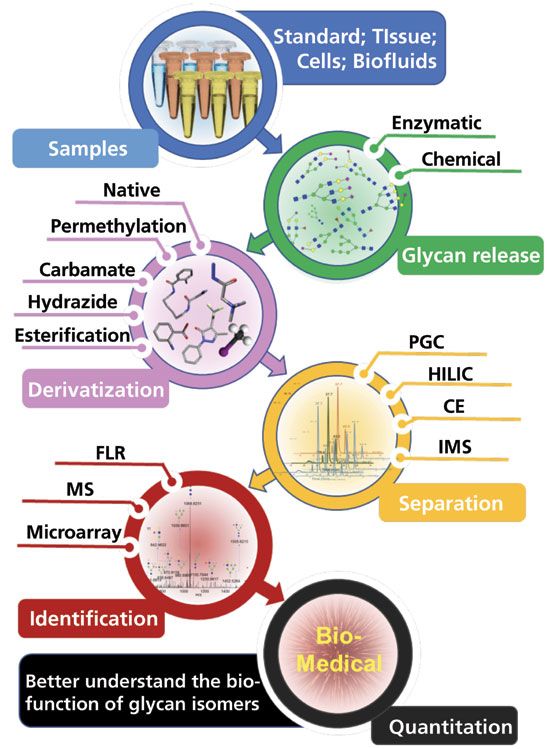
Figure 1: Schematic of isomeric glycomics strategies.
Isomeric Glycomics of Native Glycans
Lectin Array-Based Native Glycan Isomer Analysis
Based on specific binding to glycans, lectin affiliation tests are considered one of the most important tools for isomeric glycan analysis (37). Unlike MS-based methods, which can obtain all of the isomeric information in a single scan, lectin-based glycan identification extracts certain types of information from the glycan structures, such as whether the glycan is core-fucosylated or sialylated (38). When combining the results of glycan affinity with different lectins, isomeric information can be obtained. Recently, researchers performed a thorough investigation of lectin binding properties to glycan isomers (39). A library of lectin affinity was built using chemoenzymatic synthesized glycans, including sialylated, fucosylated, neutral complex, and high mannose glycan species and their isomers.
MS-Based Native Glycan Isomer Analysis
Native or reduced glycans released from samples are typically analyzed in negative mode. Native glycan analysis requires less sample preparation, which introduces minimum sample loss and variation due to human bias and errors. However, the ionization efficiency in MS is lower relative to other derivatization methods, such as reducing end labeling and permethylation. Native glycan analysis also suffers from the lack of a chromophore or fluorophore center, which therefore hinders spectroscopic detection. Despite these drawbacks, it is appealing to the researchers that native glycan analysis requires minimal sample manipulation and processing (40).
The commonly used separation method for native glycan analysis at the isomeric level is graphitic carbon chromatography. The combination of hydrophobic interaction, ionic interaction, and polarization interaction make graphitic carbon separation unique and powerful. Hayakawa and coworkers first studied the isomeric separation of reduced high mannose glycans released from ribonuclease B, and detected 12 isoforms of six structures (41). More recently, a porous graphitic carbon (PGC)-based HPLC chip was introduced to study N-glycan profiling from dried blood spots; and 150 isomeric structures were identified from 44 glycan compositions (42). Fanayan and colleagues (43) revealed the discriminate expression of α2,3-sialylation and the bisecting type GlcNAcylation between colorectal cancer (CRC) cell lines. A retention time library of reduced N-glycans on PGC was established, which included a variety of glycan species such as high mannose, sialic acid linkage isomers, and fucose positional isomers (44). This study summarized the elution rules of native glycan isomers on PGC.
Isomeric Glycomics of Derivatized Glycans
The major concern of MS-based native glycan analysis is the low ionization efficiency, which significantly reduces the sensitivity of native glycans. In addition, glycans have neither strong ultraviolet (UV) absorbance nor fluorescent emission, thus prohibiting the use of UV and fluorescence detectors. In order to overcome these drawbacks, different tags and dyes have been designed to derivatize glycans, allowing the detection of glycans by UV or fluorescence and improving the ionization for MS.
Isomeric Glycomic Studies Using Fluorescence Detectors
The common derivatization reagents for glycans, such as aniline (45), 2-Amino-pyridine (2-AP) (46), 2-aminobenzamide (2-AB) (47), and 2-aminobenzoic acid (2-AA) (48), contain a fluorescence group that allows glycans to be detected using either a UV detector or a fluorescence detector. Although there have been few improvements made to these routinely used reagents in recent years, they are still widely implemented in laboratories, due to their cost-effectiveness. In terms of fluorescent function, efforts are still underway to expand the applications of these tags when using traditional fluorescence detectors (48). Zhu and associates (48) improved the simple chemistry in aminobenzoic acid (AA) and glycan conjugation. Figure 2 depicts the strategy of using AA for further functionalization and microarray preparation. The sialylated glycan-AA conjugation isomers (6/3SL-AA) were further modified to a desired benzoic acid monoamidation product (6/3SL-AEAB) through an optimized reaction condition, then purified. AA derivatized glycans were then illustrated to enhance the bulk glycan separation, which was effectively achieved on the weak anion exchange (WAX) column or C18 column. Glycan-AEAB conjugations were further purified, and successfully utilized for a glycan microarray printing and binding assay. In addition to the use of glycan-AA to prepare neoglycoproteins, the extensive application of AA can benefit glycan production and analysis as well as glycan isomer studies.


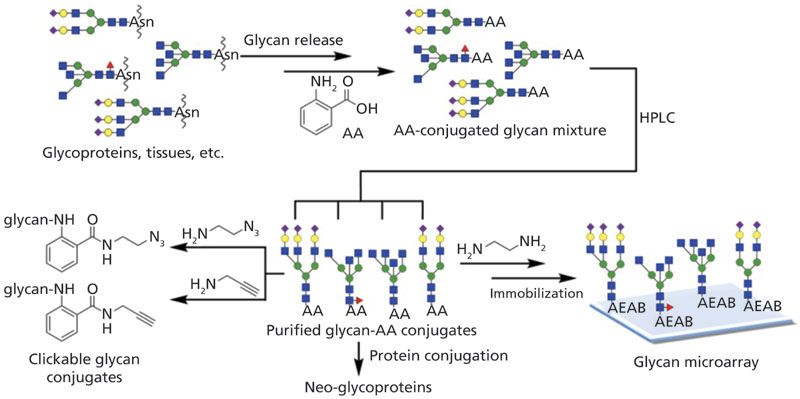
Figure 2: The strategy of the preparation of glycan–AA conjugates and versatile derivatizations of glycan–AA conjugates for further functionalization and microarray preparation (Reprinted with permission from reference 48, copyright American Chemical Society, 2018).
Although the fluorescence derivatization technique analyzes glycan isomers using a conventional fluorescence detector with a capability of stable quantitation, its application is limited by the overlapping of glycan isomer peaks and insufficient glycan isomer standards. The complete separation of all glycan isomers in a complex biological sample such as blood serum or cell lines has not yet been achieved. The lack of structural information provided by a fluorescence detector makes it hard to identify and quantify overlapping peaks. A promising solution is a combination of fluorescence and mass spectrometry. Tousiet and associates (49) investigated the combination of 2-AB as a fluorescent tag and 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM)/MeOH as a sialic acid linkage differentiator. They observed an enhancement of the isomeric separation of trisialylated glycans using an ultrahigh-pressure liquid chromatography–hydrophilic interaction chromatograph with –fluorescence detection (UHPLC-HILIC-FLR). However, the identification of isomers was still based on an MS/MS analysis. Thus, to date, fluorescence detectors have rarely been used alone in isomeric glycomic studies and have more often been used in combination with mass spectrometry.
Chemical Derivatization Enhancing MS-based Glycan Isomer Analysis
Like fluorescence detection, mass spectrometry (MS) is another glycan analysis technique that is sensitive and rich in structural information (34,35), and it has been widely used in glycomic studies. Nevertheless, native glycans have poor ionization efficiency and are highly influenced by the sample matrices and competitive ionization, thus restricting the scope of MS analysis of native glycans. These disadvantages of MS can be improved by derivatizing glycans with chemical tags that enhance ionization, or by coupling MS to separation techniques such as capillary electrophoresis (CE) and liquid chromatography (LC).
MALDI of Isomeric Glycans
Matrix-assisted laser desorption/ionization (MALDI)-MS enables the identification and quantitation of glycomics without separation (50,51). The short and straightforward sample preparation process and fast analysis speed (52) make MALDI-MS popular for glycan biomarker discovery in medical laboratories, where large numbers of samples need to be analyzed. However, the lack of a separation process with MALDI-MS has hindered isomeric glycan studies for decades, until Harvey and coworkers introduced a method for the differentiation of sialic acid linkage isomers through derivatization (53). Several derivatization methods were subsequently developed and utilized to introduce a mass difference between linkage-specific sialic acids (30,54).
Later, Wuhrer and coworkers (55) reported an optimized linkage-specific sialic acid alkylation, ethyl esterification method, which was simple, low-cost, and reproducible. Nevertheless, it has a shortcoming which cannot be ignored: Lactone derivatives are unstable, and could be completely degraded in 50 h (53). Therefore, new two-step derivatization methods were developed to overcome this issue (56). Recently, Wuhrer and colleagues (57) expanded the two-step derivatization method to MALDI-imaging. Figures 3a and 3b depict the derivatization strategy. Glycans of formalin-fixed paraffin-embedded tissues underwent the in situ two-step derivatization to introduce a 28 Da mass difference between α2,3 and α2,6 linked sialic acids. After the first step, α2,3 and α2,6-linked sialic acids were converted to their lactone and dimethylamide forms, respectively. The second step was to hydrolyze lactone and form a stable amide derivative of α2,3-sialic acid. The distributions of different sialic acid linkage isomers were altered significantly in different tissue areas, illustrated by Figure 3c, which could provide important biological information in disease studies. More recently, Nishikaze and coworkers (58) introduced an improved sialic acid linkage-specific alkylamidation (SALSA) method for MALDI-MS glycan isomeric analysis, which could offer an alternative to the aforementioned two-step derivatization approach.


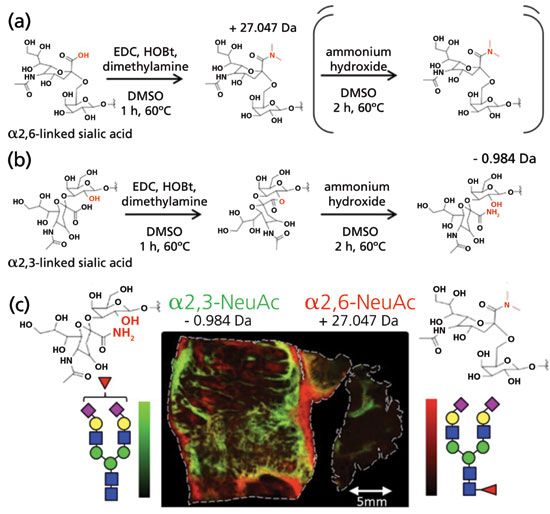
Figure 3: Reaction scheme of on-tissue N-glycan derivatization. Derivatization of N-acetylneuraminic acid which is either (a) α2,6-linked, or (b) α2,3-linked to the subterminal galactose. The reaction consists of two parts: the first, where only EDC, HOBt, and dimethylamine are present, and the second, where ammonia is also added. Under the same conditions, α2,6-linked sialic acids form a stable dimethylamide during the first step, whereas α2,3-linked sialic acids react with the neighboring galactose to form a lactone. In the second step, the lactone is hydrolyzed, and the added ammonia reacts with the carboxylic acid to form a stable amide. This results in a final mass difference between the differently linked N-acetylneuraminic acids of 28.031 Da, and enables the differentiation of α2,6- vs. α2,3-linkage, while the N-acetylneuraminic acids are stabilized for MALDI-TOF-MS analysis. (c) MALDI-imaging of on-tissue α2,6- and α2,3-linked disialylated glycan isomers. (Adapted with permission from reference 57, copyright American Chemical Society, 2016.)
Despite the rapid analysis speed and the many methods of MALDI-MS glycan isomeric analysis that have been developed during the last decade, MALDI-MS can only distinguish sialic acid linkage isomers. Other isomeric information, such as core- or branch-fucosylated isomers and different branch positional isomers, remain a significant challenge to MALDI-MS. Moreover, O-glycan isomeric studies in the MALDI-MS arena remain untouched. Thus, the use of a combination of more efficient separation techniques, such as UHPLC–MS or CE–MS, is still the most popular method of glycan isomer analysis.
CE–MS in Isomeric Glycan Study
Capillary electrophoresis analysis of glycan isomers benefits from short analysis time and peak resolution. Microfluidic chip electrophoresis has been widely used for glycomics studies due to the stability of the CE–MS interface (59). Jacobson and associates investigated methylamidated and 1-aminopyrene-3,6,8-trisulfonic-acid (APTS) derivatized glycan separation using microchip electrophoresis (60,61). Such derivatizations neutralized the negative charges introduced by sialic acids and enabled reliable quantitative analysis by LIF detection (52). Isomeric structures were identified by comparing the electrophoretic mobilities of standard glycans, exoglycosidase digestion, and migration orders. The method was later applied to investigate glycan profiling in colorectal cancer prognosis (53). In addition, Zaia and coworkers applied tandem mass tag (TMT) labeling techniques to increase the glycan migration in microfluidic chip electrophoresis (62), where the migration order of different glycan types was discussed. Glycan separation using capillary electrophoresis has been previously reviewed by Volpi and colleagues (63).
LC–MS in Isomeric Glycan Study
A commonly used glycan derivatization technique is to introduce reductive amidation on the reducing end of glycans, which leaves the glycans mainly hydrophilic. Although separation can be achieved on reversed-phase liquid chromatography due to the increase of hydrophobicity compared to native glycans, most studies have separated the reducing end-labeled glycans on hydrophilic-interaction chromatography (HILIC). Compared to native glycans, reducing end-labeled glycans benefit from higher ionization efficiency and increased compatibility with fluorescence detection. A variety of glycan labeling techniques have been recently used, such as aniline (64–66), APTS (67), procainamide (68), and 2-AB47 labeling. HILIC separation of reducing end-labeled glycans is compatible with spectroscopic detection, which is favored in quantitative analysis. However, the resolving power of separate glycan isomers is sometimes inefficient.
In addition to HILIC separation of glycan isomers, reversed-phase LC, especially C18 chromatography, has been used to study glycan isomeric profiles (69,70). However, due to the hydrophilic properties of glycans and the hydrophobic forces required to retain them on reversed-phase columns, glycan structures are frequently clustered and poorly separated (71). With an 8-aminonaphthalene-1,3,6-trisulfonic acid (ANTS) treatment, high mannose glycans could be identified (72). Alternatively, permethylation can significantly transfer glycans from hydrophilic structure into hydrophobic by converting all of the active hydrogens into methyl groups, thus enabling the possibility of separating glycans on hydrophobic column materials (73). In addition, permethylation can significantly enhance the ionization efficiency of glycans in positive mode and stabilize glycan structures, preventing fucose migration and sialic acid loss (74). The isomeric separation of permethylated N-glycans, including Man7 and sialylated structures on C18, was partially achieved under elevated temperatures (75). A review of reversed-phase LC separation of glycans was recently published by Vreeker and associates (71).
As previously mentioned in section 2.2, PGC is the most potent stationary phase for isomeric separation of derivatized glycans. Recently, Mechref and coworkers investigated isomeric separation of permethylated glycans at elevated temperatures (32). The high mannose-type glycans (Figure 4a), sialic acid linkage isomers (Figure 4b), and fucose and galactose positional isomers (Figures 4c and 4d) were successfully resolved. Although no diagnostic ions were identified in the galactose positional isomers, the ratio of MS/MS fragment peaks (Figures 4e and 4f) was distinguishable between the isomers, which was illustrated by modeling of the permethylated glycan molecules. Costello and coworkers recently achieved baseline separation of permethylated glycans that were also achieved on PGC under 75 °C, where glycan isomeric structures were assigned through electronic excitation dissociation (32,76). Glycan isoforms, including sialic acid linkage isomers and fucose/mannose compositional isomers, were confirmed by applying glycan standards and matching MS/MS fragments. N-glycan profiling of pancreatic cancer was also investigated for potential biomarker discovery (77). Methylamidated glycans were separated on a PGC column and quantified at the isomeric level, where 25 isomeric structures were identified as significant. In addition, reducing end-labeling techniques such as 12C6-/13C6-aniline labeling (78), as well as non-reducing end derivatization techniques including methylamidation methods (56,79), are also compatible with PGC-LC–MS. Despite the high separation efficiency and the stability of the column material, however, PGC columns suffer from relatively low reproducibility due to the fouling on the stationary surface. This issue can be addressed by regular regeneration of columns (80).


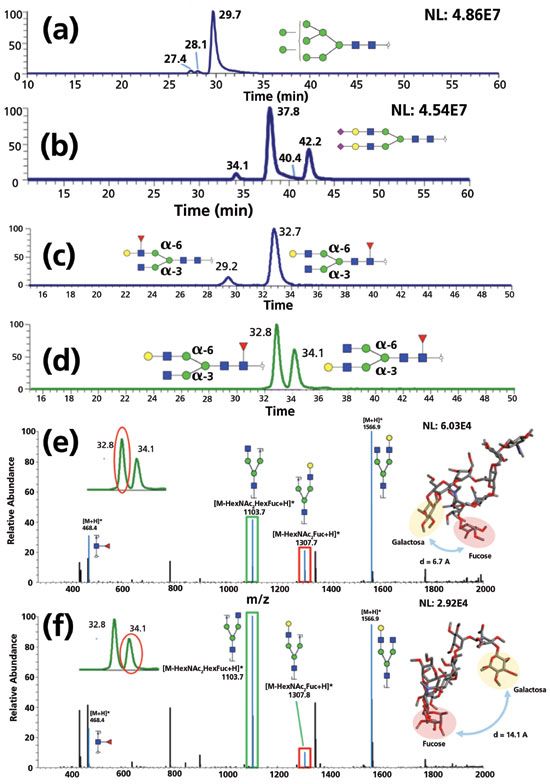
Figure 4: Isomeric separation of permethylated glycans on PGC under 75 °C. Baseline separation of (a) high mannose glycans, (b) sialylated glycans with different linkages, (c) fucose positional isomers, and (d) galactose positional isomers were achieved. The ratio of MS/MS fragment peaks (e,f) was distinguishable between the galactose positional isomers. (Adapted with permission from reference 32, copyright American Chemical Society, 2017.)
Ion Mobility Analysis of Isomeric Glycans
As a gas-phase separation technique, ion mobility mass spectrometry (IMS) has attracted more and more interest in recent years. The separation of analyte ions is achieved by movement through a drift tube, driven by an electric field and based on their masses, charges, sizes, and shapes (81,82). The similar status of analytes and instrument configuration make IMS spectacularly suitable to be coupled with MS. Drift time distributions of glycan isomers are usually transferred to a collision cross-section (CCS), which is relative to ion charge and shape under specific instrument conditions (81). Glycan isomers have different spatial structures that permit the possibility of being separated by IMS. The isomeric separation and the characterization of glycans have been demonstrated using ion mobility mass spectrometry (IMS), complementary to LC-CE–MS (83,84). Since Clemmer and coworkers (85) first separated oligosaccharide isomers using IMS, isomeric glycan analysis has developed rapidly, and glycan isomeric separations have been observed by many IMS studies (86–88). Pagel and Harvey (89) introduced a calibration protocol for traveling wave (TW) IMS using a series of sodiated glycans, which allowed glycans to be assigned by collision cross section (CCS) and also distinguished glycan isomers.
In the following studies, IMS was coupled to collision-induced dissociation (CID) mass spectrometry to acquire fragment features of glycan isomers (90). CID-IMS instrumentation was designed to acquire the fingerprint of glycan isomers, as shown in Figure 5a. Glycan isomer fragment IMS fingerprints were initially established and verified by synthetic Lewis and blood group oligosaccharides, and then applied to analyze milk oligosaccharides and human parotid tissue N-glycans. Glycan isomers could be characterized by matching biological data to a pre-established fingerprint library (Figures 5a and 5c). More recently, Harvey and associates (91) utilized the same strategy to achieve the characterization of high-mannose type glycans using CID-IMS.


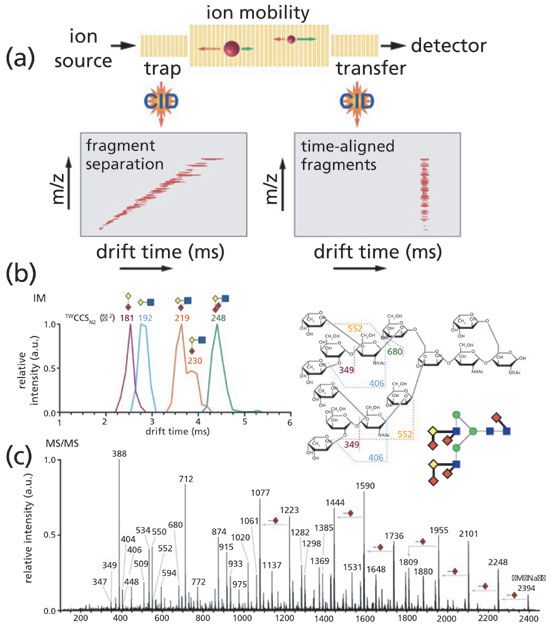
Figure 5: (a) Schematic of a TW-IM-MS instrument depicting IM separation properties of glycan fragment ions for either trap or transfer CID. (b) IM-MS/MS of the m/z 2394 [M + Na]+ N-glycan from human parotid gland with ATDs and TWCCSsN2 of diagnostic fragments shown. (c) MS/MS spectrum of m/z 2394 with an illustrated representation of the known precursor structure. Fragment assignments of the diagnostic ions are illustrated. Glycan structures are represented using the Oxford system (blue squares = GlcNAc, green circles = Man, yellow diamonds = Gal, red diamonds = Fuc). (Adapted with permission from reference 90, copyright American Chemical Society, 2017.)
Beyond the aforementioned works, additional research by Struwe and colleagues (92) and Costello and coworkers (93) have focused on an IMS database establishment of glycans. The development of such a database may increase the confidence and accuracy of glycan identification. However, as a cutting-edge technique in isomeric glycomic studies, IMS still needs to be improved. Despite its rapid separation time, which is usually MS-level, the low resolution of IMS greatly hinders the analysis of glycan isomers. The overlapping drift time distributions of glycan isomers makes IMS not as efficient as other separation techniques. Moreover, the separation of O-glycan isomers has not yet been well-investigated. Nevertheless, IMS has vast room for improvement and should be considered a useful complementary approach to CE-LC–MS.
Advancement in Analytical Techniques Facilitating Identification of Isomeric Glycans Enables a Better Understanding of Biomedical Applications
Although glycan profiling to screen for glycan biomarkers has been performed on clinical specimens (including human blood serum, cancer tissues, and urine), the existing research on glycan isomers is far from sufficient. Previous techniques have restricted efficient and reliable glycan isomer discovery from a large number of clinical samples. Fortunately, efforts to investigate potential glycan isomer biomarkers have increased in recent years with the development of the separation and identification techniques discussed above.
Clemmer and colleagues (94) observed glycan isomeric distribution alterations in serum samples from 39 liver disease patients (20 with cirrhosis of the liver and 19 with liver cancer) and 22 healthy controls using IMS. In their further study on 7 Barrett's esophagus (BE), 12 high-grade dysplasia (HGD), 56 esophageal adenocarcinomas (EAC), and 61 healthy control (NC) serum samples, the differential expressions of 46 glycan isomers could be used to distinguish the different diseases through principal component analysis (PCA) (87). However, the glycan isomer biomarker search of IMS still suffered from a low resolution, resulting in only partial separations.
Alley and Novotny (30) utilized MTT-MM/MeOH derivatization followed by permethylation to investigate different sialic acid linkages from the blood serum of 10 control and 10 breast cancer patients. The increase of α2,6 sialylation was observed in the cancer samples, which might be a potential indicator for breast cancer diagnosis. Wuhrer and coworkers (57) also observed the increase of α2,6 sialylation in colon cancer using MALDI-imaging with the in situ derivatization method.
With better isomeric separation, LC–MS has been widely used for glycan isomer quantitation in biomedical applications. Tousi and colleagues (49) observed an increased α2,6 tri-sialylated tri-antennary isomer in pancreatic cancer using HILIC-LC–MS. Lebrilla and coworkers (40) reported 38 isomers that had significant expression alteration in the serum of prostate cancer patients (n = 4) using native glycan PGC-LC–MS. Expression changes of glycan isomers were also observed in ovarian cancer (95) and pancreatic cancer (77) using a similar PGC-MS strategy. Recently, a more sensitive and efficient isomeric separation of permethylated glycan by PGC-MS at high temperature was applied to hepatocellular carcinoma (n = 10) patient sera (33). Such observations of glycan isomer expression changes in cancer demonstrate the importance of glycan isomers as potential biomarkers.
However, the study of glycan isomer biomarker discovery is still in its early stages. None of the glycan isomers have been applied in clinical diagnoses. The lack of large cohorts is one of the aspects that needs to be improved. Although more studies have focused on isomeric glycomics, the sample pools used were usually too small. A large cohort containing hundreds of thousands of samples is demanded a reliable biomarker discovery and verification. Moreover, the investigation of O-glycan isomers as biomarkers is almost nonexistent; this should receive careful consideration, given that O-glycans may participate in the bioprocess as much as N-glycans.
Concluding Remarks
The development of novel separation and characterization techniques prompts new isomeric glycomic research. Increasingly efficient and reliable strategies have been introduced for glycan isomer analysis. To date, LC–MS (assisted by a variety of glycan derivatization techniques) is the most widely used approach for isomeric glycomics. Other analyses, such as MALDI or IMS-based isomeric glycomics, can be used as supplementary or alternative methods. However, a faster and more efficient strategy, as well as a larger cohort of clinical samples, are still required to facilitate the discovery of glycan isomer biomarkers.
Acknowledgment
This work was supported by grants from The National Institutes of Health, NIH (1R01GM112490-04, 1R01GM130091-01, and 1U01CA225753-01).
References
(1) G.W. Hart and R.J. Copeland, Cell 143, 672–676 (2010).
(2) R. Zhu, L. Zacharias, K.M. Wooding, W. Peng and Y. Mechref, Methods Enzymol. 585, 397–429 (2017).
(3) X. Dong, Y. Huang, B.G. Cho, J. Zhong, S. Gautam, W. Peng, S.D. Williamson, A. Banazadeh, K.Y. Torres-Ulloa and Y. Mechref, Electrophoresis 39, 3063–3081 (2018).
(4) S.F. Tzeng, C.H. Tsai, T.K. Chao, Y.C. Chou, Y.C. Yang, M.H. Tsai, T.L. Cha, and P.W. Hsiao, Faseb. J. (29 Nov 2018). DOI: 10.1096/fj.201800687.
(5) G. de Vreede, H.A. Morrison, A.M. Houser, R.M. Boileau, D. Andersen, J. Colombani, and D. Bilder, Dev. Cell. 45, 595–605.e594 (2018).
(6) M. Oyama, Y. Kariya, Y. Kariya, K. Matsumoto, M. Kanno, Y. Yamaguchi, and Y. Hashimoto, Biochem. J. 475, 1583–1595 (2018).
(7) C. Singh, R.K. Shyanti, V. Singh, R.K. Kale, J.P.N. Mishra and R.P. Singh, Biochem. Biophys. Res. Commun. 499, 374–380 (2018).
(8) T.T. Wang, Curr. Top Microbiol. Immunol. (2019). DOI: 10.1007/82_2019_152.
(9) M. Sperandio, C.A. Gleissner, and K. Ley, Immunol. Rev. 230, 97–113 (2009).
(10) Y. Xu, W. Wu, Q. Han, Y. Wang, C. Li, P. Zhang, and H. Xu, Open Biol. 9, 180239 (2019).
(11) C. Phoomak, D. Park, A. Silsirivanit, K. Sawanyawisuth, K. Vaeteewoottacharn, M. Detarya, C. Wongkham, C.B. Lebrilla and S. Wongkham, Mol. Oncol. 13, 338–357 (2019).
(12) R.J. Sola and K. Griebenow, J. Pharm. Sci. 98, 1223–1245 (2009).
(13) R. Apweiler, H. Hermjakob and N. Sharon, Biochim Biophys Acta. 1473, 4–8 (1999).
(14) H.H. Freeze, J.X. Chong, M.J. Bamshad and B.G. Ng, Am. J. Hum. Genet. 94, 161–175 (2014).
(15) M. Van Scherpenzeel, E. Willems, and D. J. Lefeber, Glycoconj. J. 33, 345–358 (2016).
(16) R. Chandrasekaran and D.B. Lacy, FEMS Microbiol Rev. 41, 723–750 (2017).
(17) R. Jitschin, M. Bottcher, D. Saul, S. Lukassen, H. Bruns, R. Loschinski, A.B. Ekici, A. Reis, A. Mackensen, and D. Mougiakakos, Leukemia (2019). DOI: 10.1038/s41375-018-0376-6.
(18) Y. Kizuka, S. Kitazume, and N. Taniguchi, Biochim. Biophys. Acta Gen. Subj. 1861, 2447–2454 (2017).
(19) S. Schedin-Weiss, B. Winblad, and L.O. Tjernberg, Febs. J. 281, 46–62 (2014).
(20) M.L. Bermingham, M. Colombo, S.J. McGurnaghan, L.A.K. Blackbourn, F. Vuckovic, M. Pucic Bakovic, I. Trbojevic-Akmacic, G. Lauc, F. Agakov, A.S. Agakova, C. Hayward, L. Klaric, C.N.A. Palmer, J.R. Petrie, J. Chalmers, A. Collier, F. Green, R.S. Lindsay, S. Macrury, J.A. McKnight, A.W. Patrick, S. Thekkepat, O. Gornik, P. M. McKeigue, and H.M. Colhoun, Diabetes Care 41, 79–87 (2018).
(21) W. Peng, J. Zhao, X. Dong, A. Banazadeh, Y. Huang, A. Hussien, and Y. Mechref, Expert Rev. Proteomics 15, 1007–1031 (2018).
(22) A.C. Russell, M. Simurina, M.T. Garcia, M. Novokmet, Y. Wang, I. Rudan, H. Campbell, G. Lauc, M.G. Thomas, and W. Wang, Glycobiology 27, 501–510 (2017).
(23) Y. Mechref, Y. Hu, A. Garcia, and A. Hussein, Electrophoresis 33, 1755–1767 (2012).
(24) M.J. Kailemia, D. Park and C.B. Lebrilla, Anal. Bioanal. Chem. 409, 395–410 (2017).
(25) H. Guo and K.L. Abbott, Adv. Cancer. Res. 126, 281–303 (2015).
(26) K.M. Wooding, W. Peng and Y. Mechref, Curr. Pharm. Biotechnol. 17, 788–801 (2016).
(27) A. Varki, R.D. Cummings, J.D. Esko, P. Stanley, G.W. Hart, M. Aebi, A.G. Darvill, T. Kinoshita, N.H. Packer, J.H. Prestegard, R.L. Schnaar and P.H. Seeberger, Eds., Essentials of Glycobiology [Internet] 3rd ed. (Cold Spring Harbor Laboratory Press, 2015–2017).
(28) A. Yu, J. Zhao, W. Peng, A. Banazadeh, S. D. Williamson, M. Goli, Y. Huang and Y. Mechref, Electrophoresis 39, 3104–3122 (2018).
(29) R. M. Schmaltz, S.R. Hanson and C.H. Wong, Chem. Rev. 111, 4259–4307 (2011).
(30) W.R. Alley, Jr. and M.V. Novotny, J. Proteome Res. 9, 3062–3072 (2010).
(31) C.H. Chung, B. Mirakhur, E. Chan, Q. T. Le, J. Berlin, M. Morse, B.A. Murphy, S M. Satinover, J. Hosen, D. Mauro, R. J. Slebos, Q. Zhou, D. Gold, T. Hatley, D.J. Hicklin and T.A. Platts-Mills, N. Engl. J. Med. 358, 1109–1117 (2008).
(32) S. Zhou, Y. Huang, X. Dong, W. Peng, L. Veillon, D.A.S. Kitagawa, A. J.A. Aquino and Y. Mechref, Anal. Chem. 89, 6590–6597 (2017).
(33) Y. Huang, S. Zhou, J. Zhu, D.M. Lubman and Y. Mechref, Electrophoresis. 38, 2160–2167 (2017).
(34) A. Dell and H.R. Morris, Science 291, 2351–2356 (2001).
(35) Y. Mechref and M.V. Novotny, Chem. Rev. 102, 321–369 (2002).
(36) S. Zhou, L. Veillon, X. Dong, Y. Huang, and Y. Mechref, Analyst. 142, 4446–4455 (2017).
(37) S. Angeloni, J. L. Ridet, N. Kusy, H. Gao, F. Crevoisier, S. Guinchard, S. Kochhar, H. Sigrist, and N. Sprenger, Glycobiology 15, 31–41 (2005).
(38) A. Kuno, N. Uchiyama, S. Koseki-Kuno, Y. Ebe, S. Takashima, M. Yamada, and J. Hirabayashi, Nat. Methods 2, 851–856 (2005).
(39) C. Gao, M.S. Hanes, L.A. Byrd-Leotis, M. Wei, N. Jia, R.J. Kardish, T.R. McKitrick, D.A. Steinhauer. and R.D. Cummings, Cell Chem. Biol. (2019). DOI: 10.1016/j.chembiol.2019.01.002.
(40) S. Hua, H.J. An, S. Ozcan, G.S. Ro, S. Soares, R. DeVere-White. and C. B. Lebrilla, Analyst 136, 3663–3671 (2011).
(41) N. Kawasaki, M. Ohta, S. Hyuga, O. Hashimoto. and T. Hayakawa, Anal. Biochem. 269, 297–303 (1999).
(42) L.R. Ruhaak, S. Miyamoto, K. Kelly, and C.B. Lebrilla, Anal. Chem. 84, 396–402 (2012).
(43) M.K. Sethi, W.S. Hancock, and S. Fanayan, Acc. Chem. Res. 49, 2099–2106 (2016).
(44) J.L. Abrahams, M.P. Campbell, and N.H. Packer, Glycoconj. J. 35, 15–29 (2018).
(45) K. Jiang, C. Wang, Y. Sun, Y. Liu, Y. Zhang, L. Huang, and Z. Wang, J. Agric. Food Chem. 62, 7245–7254 (2014).
(46) Y. Yamaguchi, W. Nishima, S. Re, and Y. Sugita, Rapid Commun. Mass Spectrom. 26, 2877–2884 (2012).
(47) J. Zhao, S. Li, C. Li, S. L. Wu, W. Xu, Y. Chen, M. Shameem, D. Richardson, and H. Li, Anal. Chem. 88, 7049–7059 (2016).
(48) Y. Zhu, X. Liu, Y. Zhang, Z. Wang, Y. Lasanajak, and X. Song, Bioconjug. Chem. 29, 3847–3855 (2018).
(49) F. Tousi, J. Bones, W.S. Hancock, and M. Hincapie, Anal. Chem. 85, 8421–8428 (2013).
(50) A. Banazadeh, W. Peng, L. Veillon, and Y. Mechref, J. Am. Soc. Mass Spectrom. 29, 1892–1900 (2018).
(51) J. Zhong, A. Banazadeh, W. Peng, and Y. Mechref, Electrophoresis 39, 3087–3095 (2018).
(52) A.V. Everest-Dass, E.S.X. Moh, C. Ashwood, A.M.M. Shathili, and N. H. Packer, Expert Rev. Proteomics 15, 165–182 (2018).
(53) S.F. Wheeler, P. Domann, and D.J. Harvey, Rapid Commun. Mass Spectrom. 23, 303–312 (2009).
(54) X. Liu, H. Qiu, R.K. Lee, W. Chen, and J. Li, Anal. Chem. 82, 8300–8306 (2010).
(55) K.R. Reiding, D. Blank, D.M. Kuijper, A.M. Deelder, and M. Wuhrer, Anal. Chem. 86, 5784–5793 (2014).
(56) H. Li, W. Gao, X. Feng, B.F. Liu ,and X. Liu, Anal. Chim. Acta. 924, 77–85 (2016).
(57) S. Holst, B. Heijs, N. de Haan, R.J. van Zeijl, I.H. Briaire-de Bruijn, G.W. van Pelt, A.S. Mehta, P.M. Angel, W.E. Mesker, R.A. Tollenaar, R.R. Drake, J. V. Bovee, L.A. McDonnell, and M. Wuhrer, Anal. Chem. 88, 5904–5913 (2016).
(58) T. Nishikaze, H. Tsumoto, S. Sekiya, S. Iwamoto, Y. Miura, and K. Tanaka, Anal .Chem. 89, 2353–2360 (2017).
(59) E.A. Redman, N.G. Batz, J.S. Mellors, and J.M. Ramsey, Anal .Chem. 87, 2264–2272 (2015).
(60) I. Mitra, C.M. Snyder, X. Zhou, M.I. Campos, W.R. Alley, Jr., M.V. Novotny, and S. C. Jacobson, Anal. Chem. 88, 8965–8971 (2016).
(61) C.M. Snyder, W.R. Alley, Jr., M.I. Campos, M. Svoboda, J.A. Goetz, J.A. Vasseur, S.C. Jacobson, and M.V. Novotny, Anal. Chem. 88, 9597–9605 (2016).
(62) K. Khatri, J.A. Klein, J.R. Haserick, D.R. Leon, C.E. Costello, M.E. McComb, and J. Zaia, Anal. Chem. 89, 6645–6655 (2017).
(63) V. Mantovani, F. Galeotti, F. Maccari, and N. Volpi, Electrophoresis 39, 179-189 (2018).
(64) M. Mancera-Arteu, E. Gimenez, J. Barbosa, R. Peracaula, and V. Sanz-Nebot, Anal. Chim. Acta. 991, 76–88 (2017).
(65) M. Mancera-Arteu, E. Gimenez, J. Barbosa, and V. Sanz-Nebot, Anal. Chim. Acta 940, 92–103 (2016).
(66) E. Gimenez, M. Balmana, J. Figueras, E. Fort, C. de Bolos, V. Sanz-Nebot, R. Peracaula, and A. Rizzi, Anal. Chim. Acta 866, 59-68 (2015).
(67) S. Yamamoto, M. Kinoshita, T. Ikegami, and S. Suzuki, J. Chromatogr. A 1566, 44–50 (2018).
(68) S. Tao, Y. Huang, B.E. Boyes, and R. Orlando, Anal. Chem. 86, 10584–10590 (2014).
(69) R. Zhu, S. Zhou, W. Peng, Y. Huang, P. Mirzaei, K. Donohoo, and Y. Mechref, J. Proteome Res. 17, 2668–2678 (2018).
(70) L. Veillon, Y. Huang, W. Peng, X. Dong, B. G. Cho, and Y. Mechref, Electrophoresis 38, 2100–2114 (2017).
(71) G.C. Vreeker and M. Wuhrer, Anal. Bioanal. Chem. 409, 359–378 (2017).
(72) L.A. Gennaro, D.J. Harvey, and P. Vouros, Rapid Commun. Mass Spectrom. 17, 1528–1534 (2003).
(73) D.K. Williams, C.W. Meadows, I.D. Bori, A.M. Hawkridge, D.L. Comins, and D.C. Muddiman, J. Am. Chem. Soc. 130, 2122–2123 (2008).
(74) J.L. Desantos-Garcia, S.I. Khalil, A. Hussein, Y. Hu, and Y. Mechref, Electrophoresis 32, 3516–3525 (2011).
(75) S. Zhou, Y. Hu and Y. Mechref, Electrophoresis 37, 1506–1513 (2016).
(76) S. Zhou, X. Dong, L. Veillon, Y. Huang and Y. Mechref, Anal. Bioanal. Chem. 409, 453–466 (2017).
(77) Y. Liu, C. Wang, R. Wang, Y. Wu, L. Zhang, B.F. Liu, L. Cheng, and X. Liu, J. Proteomics 181, 160–169 (2018).
(78) C. Michael and A.M. Rizzi, J. Chromatogr. A. 1383, 88–95 (2015).
(79) Q. Zhang, X. Feng, H. Li, B. F. Liu, Y. Lin, and X. Liu, Anal. Chem. 86, 7913–7919 (2014).
(80) M. Pabst and F. Altmann, Anal. Chem. 80, 7534–7542 (2008).
(81) V. Gabelica and E. Marklund, Curr. Opin. Chem. Biol. 42, 51–59 (2018).
(82) M.A. Ewing, M.S. Glover, and D. E. Clemmer, J. Chromatogr. A 1439, 3–25 (2016).
(83) J. Hofmann and K. Pagel, Angew Chem. Int. Ed. Engl. 56, 8342–8349 (2017).
(84) C. Manz and K. Pagel, Curr. Opin. Chem. Biol. 42, 16–24 (2018).
(85) Y. Liu and D.E. Clemmer, Anal. Chem. 69, 2504–2509 (1997).
(86) M.D. Plasencia, D. Isailovic, S.I. Merenbloom, Y. Mechref, M.V. Novotny, and D.E. Clemmer, J. Am. Soc. Mass Spectrom. 19, 1706–1715 (2008).
(87) M.M. Gaye, S.J. Valentine, Y. Hu, N. Mirjankar, Z.T. Hammoud, Y. Mechref, B.K. Lavine, and D. E. Clemmer, J. Proteome Res. 11, 6102–6110 (2012).
(88) W. Gabryelski and K.L. Froese, J. Am. Soc. Mass Spectrom. 14, 265–277 (2003).
(89) K. Pagel and D.J. Harvey, Anal. Chem. 85, 5138–5145 (2013).
(90) J. Hofmann, A. Stuckmann, M. Crispin, D. J. Harvey, K. Pagel, and W.B. Struwe, Anal. Chem. 89, 2318–2325 (2017).
(91) D. J. Harvey, G. E. Seabright, S. Vasiljevic, M. Crispin, and W. B. Struwe, J. Am. Soc. Mass Spectrom. 29, 972–988 (2018).
(92) W.B. Struwe, K. Pagel, J.L. Benesch, D.J. Harvey, and M.P. Campbell, Glycoconj. J. 33, 399–404 (2016).
(93) R.S. Glaskin, K. Khatri, Q. Wang, J. Zaia, and C.E. Costello, Anal. Chem. 89, 4452–4460 (2017).
(94) D. Isailovic, R.T. Kurulugama, M.D. Plasencia, S.T. Stokes, Z. Kyselova, R. Goldman, Y. Mechref, M.V. Novotny, and D. E. Clemmer, J. Proteome Res. 7, 1109–1117 (2008).
(95) S. Hua, C.C. Williams, L.M. Dimapasoc, G.S. Ro, S. Ozcan, S. Miyamoto, C.B. Lebrilla, H.J. An, and G.S. Leiserowitz, J .Chromatogr. A 1279, 58–67 (2013).
Yehia Mechref, Wenjing Peng, and Yifan Huang are with the Department of Chemistry and Biochemistry at Texas Tech University, in Lubbock, Texas. Direct correspondence to: yehia.mechref@ttu.edu.













Best of the Week: AI and IoT for Pollution Monitoring, High Speed Laser MS
April 25th 2025Top articles published this week include a preview of our upcoming content series for National Space Day, a news story about air quality monitoring, and an announcement from Metrohm about their new Midwest office.




LIBS Illuminates the Hidden Health Risks of Indoor Welding and Soldering
April 23rd 2025A new dual-spectroscopy approach reveals real-time pollution threats in indoor workspaces. Chinese researchers have pioneered the use of laser-induced breakdown spectroscopy (LIBS) and aerosol mass spectrometry to uncover and monitor harmful heavy metal and dust emissions from soldering and welding in real-time. These complementary tools offer a fast, accurate means to evaluate air quality threats in industrial and indoor environments—where people spend most of their time.


