Determination of Elemental Impurities in Antacids by ICP-MS According to the Validation Protocols Defined in USP Chapters and and ICH Q3D Step 4 Guidelines
Antacids present a unique set of analytical challenges for ICP-MS. These challenges can be overcome with optimized sample preparation and instrumental analytical conditions.
This study focuses on the determination of a suite of Class 1 and 2A elemental impurities (Pb, As, Cd, Hg, Co, Ni, and V) in various over-the-counter antacid tablets by inductively coupled plasma–mass spectrometry (ICP-MS), as defined in the United States Pharmacopeia (USP) Chapter <232> and the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Step 4 Guidelines. The analysis is performed according to the recommended sample preparation procedure and validation protocols laid out in USP Chapter <233>, by optimizing both the microwave digestion procedure and the analytical methodology for these types of sample matrices that contain extremely high levels of calcium, magnesium, and other mineral components.
The United States Pharmacopeia (USP) (1) and International Conference on Harmonization (ICH) (2) have aligned the list and levels of 24 elemental impurities in pharmaceutical materials, which will be implemented on January 1, 2018. The elements of toxicological concern have been categorized into three groups based on their permitted daily exposure (PDE) levels, which are defined in USP Chapter <232> (3). To show compliance, manufacturers will need to measure the elemental contaminants by following the analytical methodology described in USP Chapter <233> (4). To supplement these new chapters, the United States Food and Drug Administration (FDA) also issued draft guidance recommendations for the pharmaceutical industry on elemental impurities in drug products that represent its current thinking on the topic from a regulatory perspective (5).
Elemental Impurities Analytical Methodology
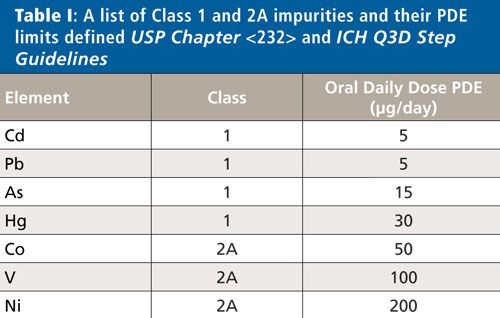
USP General Chapter <233> recommends the use of either inductively coupled plasma–mass spectrometry (ICP-MS), inductively coupled plasma–optical emission spectroscopy (ICP-OES), or any other suitable technique that meets the necessary validation protocols, for the determination of elemental impurities in finished drug products. The analytical capabilities of ICP-MS make it the most suitable technique for performing determinations of the Class 1 elements at these low levels on a routine basis-especially for drug products with a large daily dosage (>10 g/day). Among this category of medications and supplements, antacids present a unique set of analytical challenges for ICP-MS because of their extremely high calcium or magnesium content. High levels of total dissolved solids (TDS) can cause ionization suppression in the plasma as well as instrumental drift and increased maintenance because of deposition within the instrument. However, as shown in this work, these challenges can be overcome with optimized sample preparation and instrumental analytical conditions. A list of Class 1 and 2A impurities and their PDEs defined in USP Chapter <232> and ICH Q3D Step Guidelines appears in Table I.


Experimental
Sample Preparation
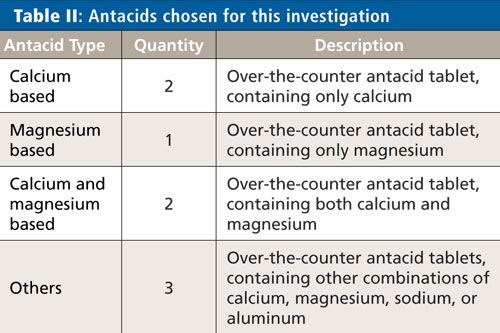
The eight antacids chosen for the evaluation represent a typical cross section of antacids available over the counter and are shown in Table II.


Microwave Digestion Procedure
USP Chapter <233> also makes suggestions on a closed-vessel or microwave digestion procedure to get solid samples into solution. A typical digestion for antacid samples uses nitric and hydrochloric acids, but because silica (SiO2) is present in some of the samples, a source of fluoride is required to dissolve the silica-typically hydrofluoric acid or tetraethyl fluoroboric acid.
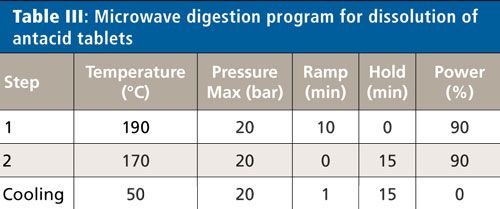
In this work, all samples were digested using a microwave digestion system (Titan MPS System, PerkinElmer Inc.) with standard 75-mL TFM (modified polytetrafluoroethylene) vessels. Approximately 3–5 g of material were crushed and homogenized. Then 0.30 ± 0.01 g of each sample was added to a digestion cup and placed in a digestion vessel, followed by 5 mL of nitric acid (70%), 1 mL of hydrochloric acid (35%), 1 mL of hydrogen peroxide (30%), and 0.5 mL of hydrofluoric acid (49%) when needed. The vessels were allowed to sit uncapped for 10 min to allow for any prereactions to occur safely before being capped and digested following the program in Table III. To evaluate the effect of the sample preparation on analyte recovery, spikes were added to the microwave vessel before the addition of acid.


When the digestion was complete, all samples were diluted with deionized water to a final volume of 50 mL, resulting in a total dilution factor of 167x with a reagent matrix of 10% HNO3, 2% HCl, and 1% HF. Calibration standards were prepared in this same matrix. To stabilize mercury, 200 ppb gold (Au) was added to each sample, standard, and blank.
Instrumentation
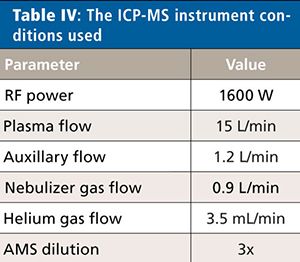
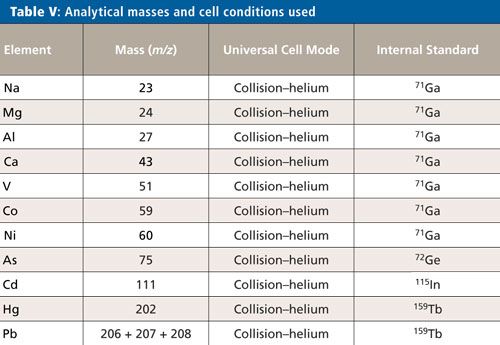
For this validation, all analyses were performed on a NexION 2000 P ICP-MS system (PerkinElmer Inc.) (6) with a SMARTintro high-throughput and high-matrix sample introduction module using standard operating conditions. This configuration offers enhanced speed of analysis through the use of an in-line flow-switching valve and provides high matrix tolerance through the use of the all matrix system (AMS), which uses aerosol dilution for enhanced matrix tolerance. Although only vanadium (V), cobalt (Co), nickel (Ni), and arsenic (As) are affected by spectral interferences originating from the sample matrix and sample preparation procedure, the ICP-MS system was operated in collision mode for all analytes and samples, providing a simple method for the analysis. The instrument conditions are shown in Table IV, and the elements and masses appear in Table V.




Calibration
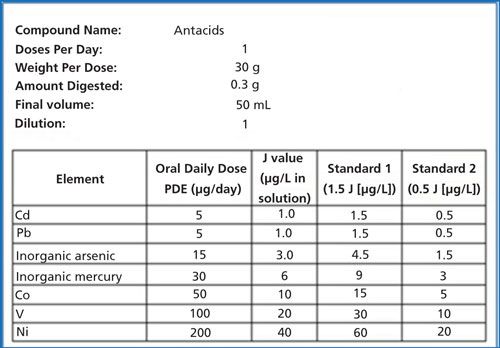
USP General Chapter <233> outlines the requirement to calibrate using a matrix-matched blank and calibration standards using concentration values of 0.5J and 1.5J. The J-value is defined as the permitted daily exposure limit for the element of interest, appropriately diluted to the working range of the instrument, after the sample preparation process is completed. In the case of antacids, large daily dosages are typical. For the eight samples in this study, the largest maximum dosage was 30 g of the antacid per day. Therefore, to test all samples using one set of calibration standards, a 30-g/day dosage was used in calculating the calibration range (0.5J and 1.5J). The elements and standard concentrations were calculated using PerkinElmer’s J Value Calculator software, as shown in Figure 1.


Figure 1: Elements and standard concentrations calculated using PerkinElmer’s J Value Calculator software.
USP General Chapter <233> also contains the following requirements for method validation:
- Accuracy: The matrix and materials under investigation must be spiked with target elements at concentrations that are 50%, 100%, and 150% of the maximum PDE. Mean spike recoveries for each target element must be within 70–150% of the actual concentrations.
- Repeatability: Six independent samples of the material under investigation must be spiked at 100% of the target limits defined and analyzed. The measured percent relative standard deviation (%RSD) must not be more than 20% for each target element.
- Ruggedness: The repeatability measurement testing procedure should be carried out by analyzing the six repeatability test solutions either on different days, either with a different instrument or by a different analyst. The %RSD of the 12 replicates must be less than 25% for each target element.
- System suitability: Standard 1 (1.5J) must be measured before and at the end of analyzing a batch of samples. The difference between the two results must be less than 20% for each target element.
Results
All quantitative sample data were less than the 0.5J standard and, as a result, were less than the target limits for the elemental impurities. Therefore, for the purposes of this study, we chose to report the method validation results for the sample that produced the greatest matrix suppression of the internal standard signal. For this reason, one of the calcium- and magnesium-based over-the-counter tablet formulations was selected because it represented the most difficult analytical challenge out of all the samples examined.
Accuracy
The accuracy data of this antacid sample is exemplified in Table VI, which shows that the predigestion spike recovery test in the sample matrix passes at all three spike levels (50%, 100%, and 150% of the target limits) with the mean spike recoveries for each target element well within the 70–150% acceptance criteria.
Repeatability
Six independently prepared samples of the Ca–Mg antacid were digested and then spiked at 100% of the target limit and analyzed. As shown in Table VII, the %RSDs for all target elements were within 2%, which is well under the 20% acceptance limit.


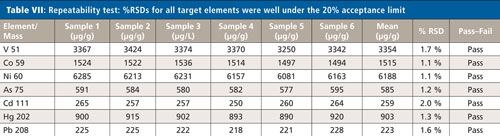
CLICK TABLE TO ENLARGE
Ruggedness
The six samples used for the repeatability study shown in Table VII were analyzed on two different days. The RSDs for these 12 measurements are all <2.5% (as shown in Table VIII), well below the method requirement of 25%.


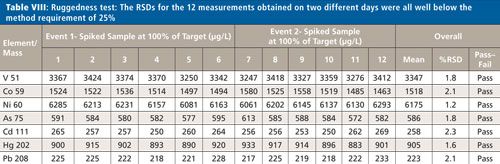
CLICK TABLE TO ENLARGE
System Suitability
To accept the sample validation data, the instrument drift must be determined by measuring Standard 1 (1.5J) at the beginning and end of the analyses. The difference between the two results for each target element was less than 6%, which is below the acceptance limit of 20%, as demonstrated in Table IX.


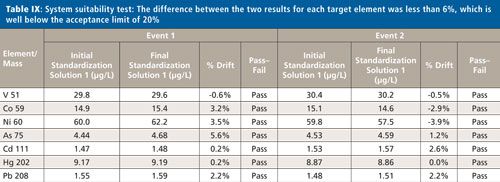
CLICK TABLE TO ENLARGE
Conclusion
This study has shown that the methodology easily handles these types of high-matrix antacid tablets. The Class 1 and 2A elemental impurity levels were well below the daily dosage PDE limits for USP Chapter <232> and ICH Q3D Step 4 Guidelines in the samples analyzed. In addition, to confirm the method validation, the most difficult antacid sample matrix was chosen and the method used for this work easily passed the acceptance criteria for the testing protocols described in USP General Chapter <233>.
References
- The United States Pharmacopeial Convention online: http://www.usp.org/usp-nf/key-issues/elemental-impurities.
- International Conference on Harmonization, ICH Q3D Step 4 - Guideline for Elemental Impurities (ICH, Geneva, Switzerland, 2014); http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q3D/Q3D_Step_4.pdf.
- General Chapter <232> “Elemental Impurities in Pharmaceutical Materials - Limits” in second supplement to United States Pharmacopeia 39-National Formulary 34 (United States Pharmacopeial Convention, Rockville, Maryland, 2016); updates published in Pharmacopeial Forum 42(2).
- General Chapter <233> “Elemental Impurities in Pharmaceutical Materials - Procedures” in second supplement to United States Pharmacopeia 38-National Formulary 33 (United States Pharmacopeial Convention, Rockville, Maryland, 2015).
- US Food and Drug Administration, Guidance for Industry: Elemental Impurities in Drug Products (FDA, Rockville, Maryland, 2016, https://www.fda.gov/downloads/Drugs/Guidances/UCM509432.pdf.
- “Thirty-Minute Guide to ICP-MS,” Perkin-Elmer Technical Note, 2017, http://www.perkinelmer.com/lab-solutions/resources/docs/TCH-30-Minute-Guide-to-ICP-MS-006355G_01.pdf.
Aaron Hineman is with PerkinElmer Inc., in Shelton, Connecticut. Direct correspondence to: aaron.hineman@perkinelmer.com









Geographical Traceability of Millet by Mid-Infrared Spectroscopy and Feature Extraction
February 13th 2025The study developed an effective mid-infrared spectroscopic identification model, combining principal component analysis (PCA) and support vector machine (SVM), to accurately determine the geographical origin of five types of millet with a recognition accuracy of up to 99.2% for the training set and 98.3% for the prediction set.



Authenticity Identification of Panax notoginseng by Terahertz Spectroscopy Combined with LS-SVM
In this article, it is explored whether THz-TDS combined with LS-SVM can be used to effectively identify the authenticity of Panax notoginseng, a traditional Chinese medicine.

