Determination of Very Low Abundance Diagnostic Proteins in Serum Using Immunocapture LC–MS/MS
Special Issues
There is growing interest in the determination of endogenous proteins in biological samples for diagnostic purposes, because a concentration increase or decrease of such proteins can allows us to monitor the state of a pathological condition such as cancer. Immunocapture LC–MS/MS analysis combines the workflow of conventional immunological assays with LC–MS analysis. This article describes typical challenges, such as cross reactivity and the mass spectrometer’s dynamic range, as well as the advantages of isoform differentiation and multiplexing.
The use of antibodies in “bottom-up” liquid chromatography–mass spectrometry (LC–MS) workflows to determine low-abundance biological active proteins in complex human samples has increased in recent years: immunocapture analysis combines the workflow of conventional immunological assays with LC–MS analysis. This article describes typical challenges, such as cross reactivity and the mass spectrometer’s dynamic range, as well as the advantages of isoform differentiation and multiplexing. Additionally, some experimental formats of immunocapture bottom-up LC–MS analysis of biological active proteins in complex human samples are discussed.
Determination of proteins in biological samples using liquid chromatography–mass spectrometry (LC–MS) is gaining much interest. An increasing amount of approved protein biopharmaceuticals, not to mention the abuse of these compounds in sports, demands the ability to determine them with a high degree of certainty in biological samples. In addition to this there is a growing interest in determination of endogenous proteins in biological samples using LC–MS for diagnostic purposes: concentration increase or decrease of such proteins allows us to monitor the state of a pathological condition, including cancer.
Generally speaking, proteins are very diverse with respect to their biological activity; however, their physicochemical properties are rather uniform. Additionally, in biological samples proteins can occur in a broad concentration range. The concentration of albumin in serum is in the medium grams-per-liter level, while the concentration of interleukin-10 in serum is in the high 10-12 g/L level. Many proteins with diagnostic value occur in the low to very low abundance range (<10-6 g/L). The diagnostic value of proteins is determined by the ability to quantify them robustly so that changes in concentration can be interpreted with good precision and accuracy. The huge span in concentrations makes this challenging because these proteins need to be measured in the presence of other endogenous proteins where the concentration in some cases is a billion (1012) times higher.
Determining Markers Using Immunological Methods
In clinical laboratories around the world, diagnostic proteins are determined using immunological methods such as enzyme-linked immunosorbent assay (ELISA) and radioimmunoassay (RIA). These are techniques that are based on the selective capture of target proteins using antibodies. After the capture and washing steps, the target proteins are quantified by measuring absorbance, fluorescence, or radioactivity. These techniques are rather simple, do not require expensive equipment, can handle many samples at the same time, and reach detection limits that allow determination of very low abundance proteins. However, a disadvantage of this approach is that the methods are prone to false results: both false positive (caused by cross reactivity) and false negative (caused by high dose hook effect) results can occur. This can lead to erroneous diagnosis with the obvious negative consequences. In addition to this, most of the assays are not able to distinguish between isoforms of the target proteins and do not allow the determination and differentiation of several target proteins in the same assay (multiplexing) (1–3).
LC–MS-Based for Targeted Protein Analysis
To cope with the disadvantages mentioned previously, the use of LC–MS in target protein analysis has gained more interest. There are two main strategies to determine proteins using LC–MS: the top-down approach and the bottom-up approach. In the top-down approach, proteins are analyzed intact. Although feasible, this approach does not allow determination of very low concentrations. A bottom-up approach (4) is chosen for quantification most often (see Figure 1). This approach is based on the enzymatic digestion (mostly trypsin) of the target protein into lesser peptides. When trypsin is used as an enzyme for this purpose, the resulting peptides will mainly be doubly charged and will give rise to good peak intensities in the mass spectrometer. The peak intensity of a tryptic peptide is higher than that of the undigested protein, which makes the bottom-up approach attractive because it gives good detection limits. As a single protein will give rise to many tryptic peptides and hence a complex chromatogram, it is therefore important to choose the right tryptic peptides for quantification.


Figure 1: Bottom-up approach in LC–MS-based analysis. Proteins are digested into peptides. Specificity of these peptides for its parent protein can be evaluated using Basic Local Alignment Search Tool (BLAST: http://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins). LC–MS/MS analysis will allow peptide identification and quantification.
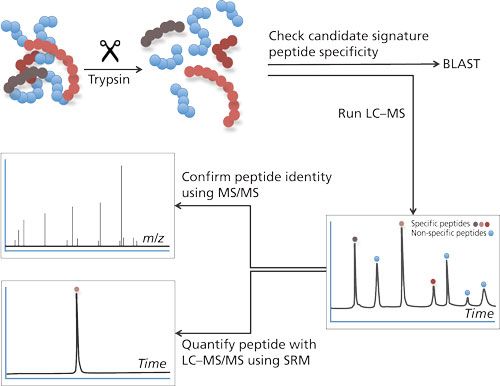
The amino acid sequence of the tryptic peptide chosen for quantification needs to be highly specific; in other words, no other protein in the sample should produce a peptide with the same amino acid sequence. Such specific peptides are designated as signature peptide or proteotypic peptide: the parent protein can be determined by means of this signature peptide. This peptide needs to be specific and, preferably, gives a high peak intensity, has good chromatographic properties, is the result of a full digest (no cleavage sites left in the peptide), and is reproducible.
The value of the mass spectrometer becomes clear when the identity of the signature peptide needs to be confirmed and the concentration needs to be determined. Confirmation of the identity is performed by both mass (m/z value) and fragmentation pattern. For this purpose most tandem mass spectrometers are suited (5). Quantification is preferably performed on a triple quadrupole mass spectrometer in the selected reaction mode (SRM) mode because this is still the most sensitive way to determine peptides; the mass spectrometer is highly selective in both confirmation and quantification of the signature peptide-false positives as a result of other cross reacting proteins will be avoided (6).
Challenges When Analyzing Proteins in Serum
To measure a target protein in a buffered solution with LC–MS after a bottom-up pre-treatment is often a straightforward procedure. Detection limits down to the femtogram level can be reached (of course, this depends on the sensitivity of the instrument used) without doing specific cleanup or enrichment (or both). This ideal situation changes rapidly when the same target protein needs to be determined in a biological matrix like serum or urine. Without any special treatment, such as clean-up or enrichment, a serum sample will generate a huge amount of peptides as well as many different peptides when tryptic digestion is performed. This causes a great deal of trouble for the mass spectrometer because its ability to determine low concentration and high concentration simultaneously is limited by a limited dynamic range. In addition, ion suppression occurs in the electrospray when the signature peptide of the target protein co-elutes with other, high concentration, peptides. To be able to perform robust quantitative analysis of target proteins in the very low abundance range using LC–MS sample preparation, as well as trypsinization, is required.
Sample Preparation Techniques for Target Protein LC–MS Analysis
There are several techniques described to perform sample cleanup and enrichment. The choice of the sample preparation strategy depends very much on the concentration of the target protein. In some cases the concentration of the target protein is so high that direct injection of a digested serum sample in the LC–MS could be performed. When the concentration becomes lower, sample cleanup, such as depletion (7), precipitation and restricted access material (RAM) (8), affinity cleanup (9–11), and molecular imprints (12) is used.
Affinity cleanup can be performed by either using antibodies, which have a high affinity against the parent protein, or by using antibodies, which have a high affinity against the signature peptide (SISCAPA) (13).
In this overview, the focus is on the use of antibodies directed against the whole protein in the sample cleanup and enrichment. The article is meant to provide an introduction to the experimental approach of immunocapture LC–MS, rather than being a comprehensive review of all work done in the field. Immuno-capture LC–MS has fewer disadvantages compared to immunological assays. This is because mass spectrometric detection is much more selective than the immunological assays: a false positive analysis in an immunological assay, caused by cross reactivity, will not be false positive in an LC–MS analysis: the signature peptide for the target protein will not be present in the cross-reacting protein and will not therefore generate a signal.
There are several ways to use antibodies to enrich target proteins. In general, the principle is the same: the antibody is immobilized on a solid support, which can be the wall of a 96-well plate (14), on sorbents packed in a small column (15), or on small magnetic beads (16). The biological sample is incubated with the antibodies thereby allowing the target protein and the antibodies to interact. After this incubation, the excess of biological sample is washed away. After some repetitive washing steps the target protein is either eluted and digested, or digested directly into its tryptic peptides before final analysis.
How Does Immunocapture Function in a 96-Well Format?
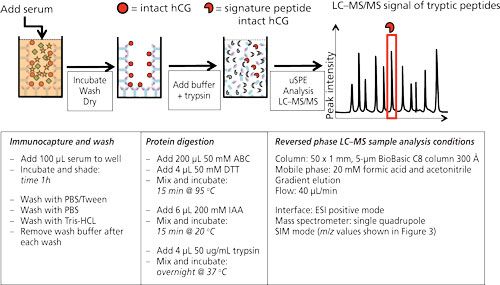
Figure 2 shows the workflow as well as the typical experimental conditions of targeted LC–MS analysis on human chorionic gonadotropin (hCG) using antibodies immobilized on the walls of a 96-well plate. hCG is used as a marker to diagnose ovarian or testicular cancer. It is also on the World Anti-Doping Agency’s list of prohibited substances. It is therefore of interest to be able to measure this protein. In the procedure shown antibodies (anti-hCG) are immobilized by incubating a solution in the wells. After blocking and washing the wells, they are ready to use. In this example serum is applied to the wells and allowed to incubate for 1 h. After washing and drying, ammonium bicarbonate buffer is added to the wells and a standard procedure to reduce, alkylate, and digest is performed. In other words, the protein is digested in the well without prior elution of the target protein from the antibody. Since the volume of the digestion buffer must be the same as the volume of the applied serum sample, only cleanup is achieved and no enrichment. The latter might be achieved by a simple μSPE step using reversed-phased material (C8 or C18) to retain tryptic peptides (14).


Figure 2: 96-well plate format for the immunoextraction of hCG from serum. Anti-hCG antibodies are immobilized on the wall of a microtiter plate. hCG in serum will be captured by these antibodies and remain in the well after extensive washing. Trypsin will digest the captured hCG, yielding a clean sample, after which μSPE can be injected into the LC–MS system. Typical experimental conditions are given in the lower part of the figure [from (14)].
A similar procedure is described for biomarkers like ProGRP (17) (small cell lung cancer), PSA (18) (prostate cancer), and protein therapeutics (19).
In the example from Figure 2 the amount of hCG is determined by measuring the amount of its signature peptide. Although hCG is often mentioned as one protein, it consists of an α-chain and a β-chain. The α-chain occurs in hCG and in other proteins. However, the β-chain is specific for hCG, and thus does not occur in other proteins. Aside from the fact that hCG is a heterodimeric protein, variations in the β-chain occur, giving rise to several isovariants. These isovariants have mainly the same amino acid sequence but there are also differences. A conventional immunological method would allow determination of the amount of total hCG, but would not be able to differentiate between these variants. One of the biggest advantages of using immunocapture LC–MS is that it can differentiate between various isovariants in a single run. Figure 3 shows a table of the amino acid sequence of four of these β-chain isovariants of hCG. Each isovariant has its own signature peptide with MS/MS fragments and a specific retention time. From the chromatogram in the same figure it can be seen that each isovariant produces its own peak. When using the procedure shown in Figure 2 on urine and serum, both intact hCG and some of the isovariants are detected (Figure 4).


Figure 3: Four of the isovariants of hCG. All these have their specific signature peptides with their specific m/z value and retention behavior. Within a single analysis these four isovariants can be monitored simultaneously (14).
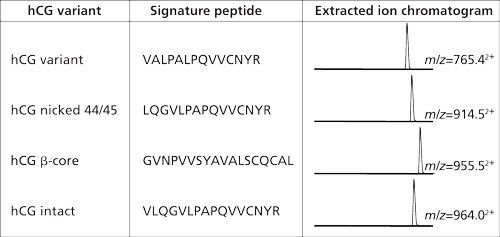


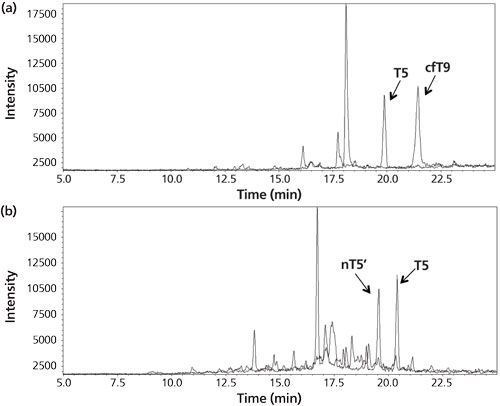
Figure 4: (a) Chromatogram for the analysis of the urine of a woman pregnant in the third month. The peaks correspond to the signature peptides for intact hCG (T5) and core fragment hCG (cfT9). (b) Chromatogram from the analysis of a cancer patient serum sample. The peaks correspond to the signature peptides for intact hCG (T5) and hCG nicked 44/45 (nT5’). Adapted and reproduced with permission from reference 14.
Although very clean chromatograms are obtained using antibodies immobilized on the walls of a 96-well plate, there is a drawback. Since the volume of the well is approximately 200–250 μL, it is not possible to use sample volumes higher than this. In some cases it is advantageous to use high volumes because the concentration and therewith the amount of target protein can be very low in the biological sample. This may be circumvented by the immobilization of antibodies on the surface of magnetic beads.
Immunocapture Using Antibodies Immobilized on Magnetic Beads
Magnetic beads are small spherical beads with a uniform size distribution (mainly 2.8 μm). The immunoactive magnetic beads can be pipetted into a serum sample of which the volume can be up to 1 mL (or even higher). Extraction of the beads (and therewith the target protein) is performed using a magnet (16, 20–22). Figure 5 shows the extraction of ProGRP (23). Experimental conditions, workflow, and a typical result are included in this figure. ProGRP is a highly selective and sensitive biomarker for small cell lung cancer. It is not only used to diagnose the disease, but also to monitor the success of the treatment.


Figure 5: Immunocapture of ProGRP using anti-ProGRP antibodies coupled to magnetic beads. Large sample volumes can be used in the capture of ProGRP. Using a magnet the beads, with the captured ProGRP, can be retained. After extensive washing trypsin can be used to digest ProGRP. This digested sample can be injected into the LC–MS system. This approach allows determination of the total amount of ProGRP as well as quantification of ProGRP isoform 1 and ProGRP isoform 3. Internal standard (IS) for total ProGRP is included in this method (23). Typical experimental conditions are given in the lower part of the figure. The chromatogram shown is that of a spiked serum sample.
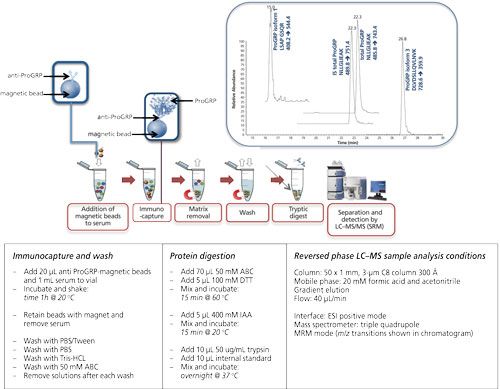
As can be seen from this figure, the principle of the extraction is comparable to that of the 96-well plate extraction. The biggest differences are the volume of the sample, which can be increased, and also the enrichment of the target protein. It is possible to start with 1 mL of serum sample from which the extraction is performed. This is done by gently shaking for 1 h (at room temperature [RT]). The magnet is used to retain the beads when the sample is removed. After washing the beads, the digestion can be performed in a much smaller volume such as for 80 μL, which would give a potential enrichment of 12.5. An additional solid-phase extraction (SPE) step would provide an even higher enrichment. In this example digestion is performed directly on-beads, meaning that no elution of the target protein is performed.
In the case of ProGRP the possibility of using larger sample volumes is of great importance: the concentration of this protein usually does not exceed 60 pg/mL and increases in the case of pathology. With 96-well based immunocapture the amount of protein that could be captured was simply too low to be determined. However, using a whole milliliter of serum allows a much better detection limit, making it possible to determine even non-pathological levels.
In addition, in the case of ProGRP, isoforms also occur. The conventional immunological assays will only be able to determine the amount of total ProGRP; with the immuno-MS approach some isoforms can be differentiated. Figure 6 shows a table of the amino acid sequence of the three known isoforms of ProGRP. With the chosen approach one can determine both ProGRP isoform 1 and isoform 3 (for isoform 2, no signature peptide was identified) in addition to the amount of total ProGRP through a signature peptide, which is common for all three isoforms.


Figure 6: Amino acid sequence of three ProGP isoforms. Each isoform contains the sequence NLLGLIEAK (in bold), which is used to determine the total amount of ProGRP. Isoform 1 and 3 each have a detectable signature peptide (LSAPGSQR for isoform 1 and DLVDSLLQVLNVK for isoform 3) that allows differentiation.

Besides immobilization of antibodies on magnetic beads, there are other formats described in the literature: Griffiths and colleagues used agarose beads as a solid support for factor VIII inhibitor analysis with MS (24); Lin and colleagues used latex beads as solid support for the immune-precipitation of several biomarker candidates (25); Sherma and colleagues used affinity pipette tips to enrich proteins before analysis (26); and Baily-Chouriberry and colleagues used anti-EPO monoliths for enrichment and subsequent MS analysis (27).
Simultaneous Immunocapture and Determination of Multiple Diagnostic Markers
Another advantage, as well as a better insight into the distribution of several isoforms, is that immune-extractions using magnetic beads can be performed without considerable volume loss. The patient sample can be reused for other analyses: After extraction of the target protein, all the other components are still present in the serum sample. Therefore, additional analyses on the same sample can be performed. Using magnetic beads, these additional analyses can be combined in a single sample cleanup–enrichment and analysis (28,29).
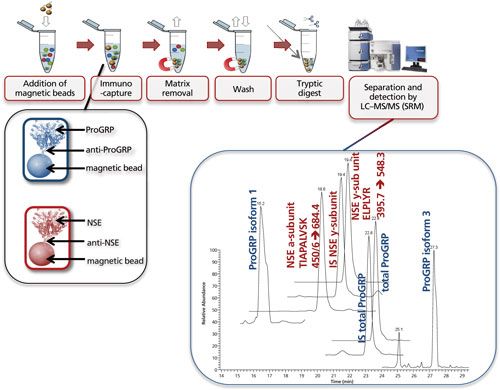
Figure 7 shows the multiplexed determination of both ProGRP and neuron-specific enolase (NSE). Experimental conditions, workflow, and a typical result are included in the figure caption. This multiplex is based on merging two existing validated methods for the determination of ProGRP (23) and NSE (20). By using a mix of two types of modified magnetic beads (one coated with anti-ProGRP and one coated with anti-NSE), these proteins can be extracted from a serum sample simultaneously. One reason to determine both proteins is that NSE, like ProGRP, is a diagnostic marker for small cell lung cancer. In other words, by doing this multiplex determination, a more robust diagnosis can be performed. As this dual extraction gives exactly the same results as two separate extractions, the time gain is also considerable. The method shown here was used to determine both ProGRP, its isoforms, and the NSE isoforms in samples from cancer patients. There was good correlation between data obtained from the conventional immunological method and the multiplex LC–MS method. In addition, an isoform distribution was determined (28). The clinical relevance of this remains to be explored, however, one can imagine that additional diagnostic proteins can be added to this test panel, potentially giving a differentiated diagnosis of the patients’ state.


Figure 7: Multiplexing: simultaneous immunocapture of ProGRP (and its isoforms) and NSE (and its various subunits) using a mixture of anti-ProGRP antibodies coupled to magnetic beads and anti-NSE antibodies coupled to magnetic beads. After the beads are added to the sample, the procedure is similar as in Figure 5. LC–MS analysis allows simultaneous determination of total ProGRP, ProGRP isoform 1, ProGRP isoform 3, NSE α-subunit, and NSE γ-subunit. Internal standards (IS) for total ProGRP and NSE γ-subunit are included in this method (28). Typical experimental conditions for such an experiment are given in the lower part of Figure 5. The only difference is that besides 20 μL of anti ProGRP beads, 20 μL of anti NSE beads is also added to the serum sample. The chromatogram shown is that of a spiked serum sample. m/z transitions for the NSE subunits are shown in the chromatogram.
Sequential Immunocapture for Differentiation of Heterodimeric Markers
The possibility of reusing the patient sample after the initial immunocapture step also opens up the potential for sequential analysis of biomarkers that cannot be separated by LC–MS/MS. An example of this is differentiation between intact hCG and its free hCGβ-subunit. This could be of interest both in cancer diagnosis and doping analysis. As mentioned earlier, hCG is a dimer consisting of an α- and a β-subunit, where the β-subunit is specific for hCG and therefore the signature peptide used for quantification must be derived from the β-subunit. This makes differentiation and quantification of intact hCG and the free β-subunit difficult using a single immunocapture step. Currently, sequential immunocapture is described for differentiation of intact hCG, hCGβ and hCGβ-core in urine (only), by first depleting urine for the free hCGβ-variants using an antibody selective to the β-subunit before extracting the remainder variants using an antibody able to capture all hCG variants (21). The captured intact and free variants are further processed and analyzed in two separate runs.
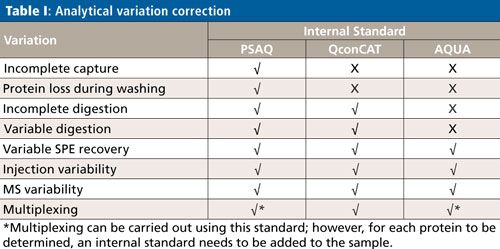
Calibration Strategies for Accurate Protein Determination
Although the subject of calibration is not within the scope of this overview, it is important to be aware that quantification in targeted protein analysis is not as straightforward as quantification of small molecules. To be able to perform an accurate quantitative analysis of the target protein (or proteins), there are several internal standard calibration strategies that can be applied: AQUA (Absolute QUAntification), PSAQ (Protein Standard for Absolute Quantification) and QconCAT (concatamer of standard Q-peptides). The goal of these strategies is to eliminate variations in the analytical process, which comprises capture, cleanup, digestion, SPE, and an LC–MS step. Table I shows which of the analytical variations are eliminated per standard calibration strategy.


Conclusions
Combining immunocapture with LC–MS has resulted in an approach with sensitivity in the same range as immunological assays. However, it can be said that detection limits are still slightly better for the immunological assays, although, with the current development in MS, there will soon no longer be any difference. Disadvantages of the immunocapture LC–MS approach compared to the immunological methods are high operational costs and the need for highly trained personnel, both as a result of the LC–MS instrument. One might argue that this approach is only feasible for exploratory biomarker research and less suited for low-cost routine determination. On the other hand, immunocapture LC–MS allows various isoforms to be differentiated between without using isoform specific antibodies. This potentially allows for more differentiated diagnosis. The possibility of determining many biomarkers at the same time in the same analysis not only saves time, but also gives a more robust diagnostic profile of a patient. Although immunocapture LC–MS also uses antibodies in the sample cleanup, it is much less, or not at all, prone to false positive results because the mass spectrometric detection is very specific. Efforts are being made to perform LC–MS analysis without the use of the immunocapture as cleanup. Although protein determination is possible, it does not come close to the detection limit needed for low-abundance biomarker determination.
All in all, immunocapture LC–MS is potentially a very powerful tool in the field of diagnostics.
References
- S. Dodig, Biochemia Medica19, 50–62 (2009).
- N. Jassam, C.M. Jones, T. Briscoe, and J.H. Horner, Ann. Clin. Biochem. 43, 314–317 (2006).
- A.N. Hoofnagle and M.H. Wener, J. Immunol. Methods347, 3–11 (2009).
- R. Aebersold and M. Mann, Nature422, 198–207 (2003).
- A. Doerr, Nat. Meth. 10, 23 (2013).
- P. Picotti and R. Aebersold, Nature Methods9, 555–566 (2012).
- L. Anderson and C.L Hunter, Mol. Cell. Proteomics5, 573–588 (2006).
- B. Winther, P. Moi, E. Paus, and J.L.E. Reubsaet, J. Sep. Sci.30, 2638–46 (2007).
- D. Nedelkov, Expert Rev. Proteomics3, 631–40 (2006).
- B.L. Ackermann and M.J. Berna, Expert Rev. Proteomics4, 175–86 (2007).
- W.H. Dunham, M. Mullin, and A.C. Gingras, Proteomics12, 1576–90 (2012).
- C. Rossetti, A. Abdel Qader, T.G. Halvorsen, B. Sellergren, and L. Reubsaet, Anal. Chem.86, 12291–12298 (2014).
- N.L. Anderson, A. Jackson, D. Smith, D. Hardie, C. Borchers, and T.W. Pearson, Mol. Cell. Proteomics8, 995–1005 (2009).
- H. Lund, S.B. Torsetnes, E. Paus, K. Nustad, L. Reubsaet, and T.G. Halvorsen, J. Proteome Res.8, 5241–5252 (2009.).
- T. Kosaka, R. Okuyama, W. Sun, T. Ogata, T. Harada, and K. Araki, Anal. Chem. 77, 2050–2055 (2005).
- H. Lund, K. Løvsletten, E. Paus, T.G. Halvorsen, and L. Reubsaet, Anal. Chem. 84, 7926–7932 (2012).
- B. Winther, M.S. Nordlund, E. Paus, L. Reubsaet, and T.G. Halvorsen, J. Sep. Sci. 32, 2937–2943 (2009).
- V. Kulasingam, C.R. Smith, I. Batruch, A. Buckler, and D.A. Jeffery, J. Proteome Res.7, 640–647 (2008).
- W. Yang, R. Kernstock, N. Simmons, and A. Alak, Bioanalysis 7, 307–318 (2015).
- S.B. Torsetnes et al., J. Chromatogr. B Analyt. Technol. Biomed. Life Sci.929, 125–132 (2013).
- G.A. Woldemariam and A.W. Butch, Clin. Chem. 60, 1089–1097 (2014).
- M. Vogel et al., Anal. Chem. 86(24), 12014–12021 (2014).
- S.B. Torsetnes, M.S. Nordlund, E. Paus, T.G. Halvorsen, and L. Reubsaet, J. Proteome Res.12, 412–420 (2013).
- A.E. Griffiths, W. Wang, F.K. Hagen, and P.J. Fay, J. Thromb Haemost. 9, 1534–1540 (2011).
- D. Lin, W.E. Alborn, R.J.C. Slebos, and D.C. Liebler, J. Proteome Res. 12, 5996–6003 (2013).
- N. Sherma et al., Proteome Sci.12, 1–12 (2014).
- L. Bailly-Chouriberry et al., Analyst. 137, 2445–2453 (2012).
- S.B. Torsetnes, M.S. Levernæs, M.N. Broughton, E. Paus, T.G. Halvorsen, and L. Reubsaet, Anal. Chem. 86, 6983–6992 (2014).
- R. Villar-Vázquez et al., Proteomics16(8), 1280–1290 (2016).
Léon Reubsaet is professor in pharmaceutical analysis at the School of Pharmacy, University of Oslo, Norway. He works with mass spectrometry as a diagnostic tool for the determination of protein biomarkers in complex biological samples. Improvement of sample handling, sample preparation, and bottom-up LC–MS strategies are his main focus. Trine Grønhaug Halvorsen is an associate professor in pharmaceutical analysis at the School of Pharmacy, University of Oslo, Norway. Her research interests include development of new strategies for analysis of protein biomarkers in biological matrices by LC–MS/MS with a main focus on novel sampling materials and sample preparation of low-abundance markers.














High-Speed Laser MS for Precise, Prep-Free Environmental Particle Tracking
April 21st 2025Scientists at Oak Ridge National Laboratory have demonstrated that a fast, laser-based mass spectrometry method—LA-ICP-TOF-MS—can accurately detect and identify airborne environmental particles, including toxic metal particles like ruthenium, without the need for complex sample preparation. The work offers a breakthrough in rapid, high-resolution analysis of environmental pollutants.






