Development of a Fast LC–MS-MS Screen for Common Drugs of Abuse as an Alternative to Immunoassay Screening
Special Issues
Enzyme immunoassay (EIA) is a conventional drug screening technique, but it can be limited by cross-reactivity that can lead to high false positive rates.
Enzyme immunoassay (EIA) is a conventional drug screening technique, but it can be limited by cross-reactivity that can lead to high false positive rates. To improve the selectivity and efficiency of a urine screen for drugs of abuse, mass spectrometry (MS) was investigated as an alternative. An existing multianalyte MS method for amphetamine, benzoylecgonine, 3,4-methylenedioxyamphetamine (MDA), methylenedioxyethylamphetamine (MDEA), 3,4-methylenedioxymethamphetamine (MDMA), methamphetamine, and phencyclidine (PCP) was modified to optimize for speed and to be a competitive screen.
Recently, many new direct-analysis mass spectrometry (MS) techniques have been developed primarily for screening purposes, such as high-throughput systems (1–10), laser diode thermal desorption (LDTD) (11–19), direct analysis in real time (DART, JEOL) (20–32), and desorption electrospray ionization (DESI) (20,33–44). These techniques have proven to be time efficient and useful in drug sample analysis (2,6,10,13,15,16,20–23,26,28,32,34,35,37,41,42,44). However, all of these techniques require special and expensive equipment in addition to a typical MS system that most often is purchased in tandem with a liquid chromatography (LC) system. Another downside is that these techniques can require a specific sample "holder." For example, one high-throughput approach currently only uses 96- or 384-well plates, and the LDTD system has its own special plate as well, which may not be ideal for small-volume assays because the need for automated liquid handling becomes a necessary additional expense. This additional equipment cost for smaller laboratories may not always be feasible. Therefore, a less-expensive and time-efficient LC–MS-MS alternative screening technique was investigated.
Using conventional LC system components and short columns or commercially available guard columns, a rapid on-line sample preparation and injection (ROSPI) method was developed. ROSPI was developed using the following guidelines:
- The method cycle time had to be less than 2 min and ideally ~1 min.
- Short columns were used to help reach the method cycle time and maintain system pressures.
- Complete separation of the peaks or interferences was not necessary.
- Only one transition was used per analyte or internal standard (IS) to ensure a sufficient number of points across the peak.
- The number of IS compounds was minimized.
- Two-point calibration near the cutoff was used for semiquantitation to determine the positivity of samples.
A current LC–MS-MS confirmation method that included commonly abused drugs was used as a proof of concept for the ROSPI method. This method included seven analytes, shown in Figure 1: amphetamine, benzoylecgonine (a cocaine metabolite), 3,4-methylenedioxyamphetamine (MDA), methylenedioxyethylamphetamine (MDEA), 3,4-methylenedioxymethamphetamine (MDMA), methamphetamine, phencyclidine (PCP). The method also included five IS compounds: amphetamine-D5, benzoylecgonine-D3, MDMA-D5, methamphetamine-D5, and PCP-D5. Typically, the compounds in this method would be screened by four different enzyme immunoassay (EIA) tests: amphetamines (amphetamine and methamphetamine), MDMA (MDA, MDEA, and MDMA), benzoylecgonine, and PCP. However, with MS all of these compounds can be screened with a single injection in less than 2 min. Furthermore, implementing with existing equipment and less expensive guard columns affords rapid screening and improved cost efficiency. Using the aforementioned goals, a ROSPI method was developed for these drugs and compared to EIA for selectivity and sensitivity using authentic patient samples.


Figure 1: Structures of drugs analyzed.
Experimental
Materials
Amphetamine, amphetamine-D5, benzoylecgonine, benzoylecgonine-D3, MDA, MDEA, MDMA, MDMA-D5, methamphetamine, methamphetamine-D5, PCP, and PCP-D5 controls were all purchased from Cerilliant. LC solvents, acetonitrile, and formic acid were purchased from Fisher Scientific and methanol was purchased from EMD Millipore. Drug-free, normal human urine was purchased from UTAK Laboratories. All water was acquired from an in-house Barnstead Nanopure purification unit from Thermo Scientific. Polypropylene 700-μL vials and snap caps with septa were purchased from VWR International.
Samples were run on an Agilent 6460 QQQ mass spectrometer with an Agilent 1200 Series Binary Pump, a column compartment with a CTCPal autosampler, and an Agilent Jet Stream electrospray ionization (ESI) source. Columns were purchased from Phenomenex and included a 30 mm × 3.00 mm, 2.6-μm dp, 100-Å Kinetex C18 column for the ROSPI method and a 50 mm × 3.00 mm, 2.6-μm dp, 100-Å Kinetex C18 column for the confirmation method. Both columns were used in tandem with a Phenomenex SecurityGuard Ultra C18 guard column and holder. Data were analyzed on Agilent MassHunter Quantitative Analysis software.
Methods
Aliquots of drug-free urine were fortified with amphetamine, benzoylecgonine, MDA, MDEA, MDMA, methamphetamine, and PCP at various concentrations for the calibration curves and a quality control (QC). For the confirmation method, all compounds except PCP were prepared at concentrations of 25, 50, 100, 150, 500, 1000, and 2500 ng/mL. PCP was prepared at additional concentrations of 4 and 10 ng/mL and excluded from the 2500-ng/mL point. For the screen method, all compounds except PCP were prepared at concentrations of 100, 150, and 300 ng/mL; PCP was prepared at concentrations of 25, 50, and 150 ng/mL. All samples were diluted 20× with 500-ng/mL amphetamine-D5, benzoylecgonine-D3, MDMA-D5, methamphetamine-D5, and PCP-D5 in water and centrifuged before a 20-μL injection onto the column. The gradient for the confirmation method was 1.87 min with a 3.12 min cycle time. The gradient was improved to 0.8 min with a ~1.7 min cycle time for the screening method. The flow rate was the same for both methods at 0.6 mL/min and the solvents used were 0.1% formic acid in water for solvent A and 50:50 acetonitrile–methanol + 0.1% formic acid for solvent B. Additionally, a wash 1 solvent of 50:50 methanol–water and a wash 2 solvent of 100% methanol were used on the CTCPal system. The additional cycle time for the methods is related to the washing and injection cycle of the CTCPal system.
In the confirmation method, two multiple reaction monitoring (MRM) transitions were used for every analyte, including the IS compounds. In the screening method, the quantifier ion transition, or the most abundant transition, was used for every analyte, including the IS compound. For the simplicity of the screening method, only benzoyl-ecgonine-D3 was used as the IS compound in the processing method, even though all IS compounds were present in the sample. This was done to demonstrate the ability to decrease cost for the screen by using a single IS compound instead of five. Data were acquired in positive ESI mode and all MS conditions remained the same for both methods.
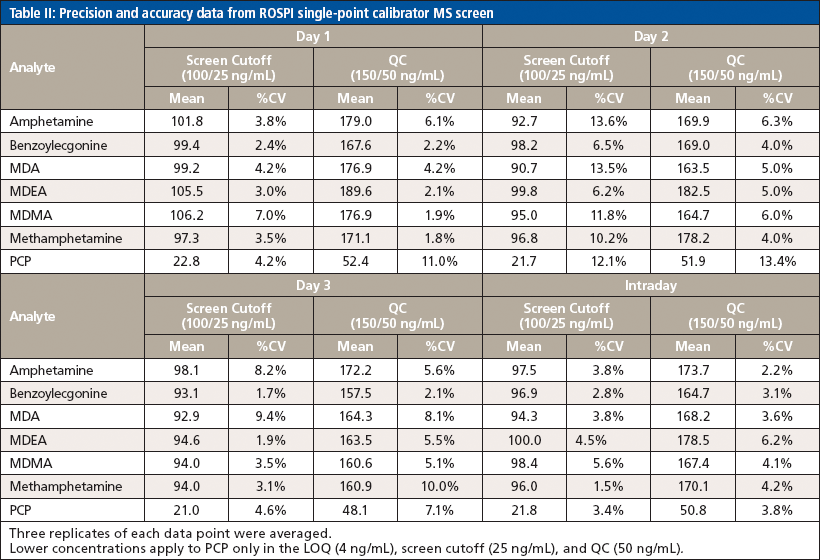
Because the confirmation method was previously validated, validation experiments were performed only for this screening method and included limits, precision and accuracy, and patient comparison. For the limits experiment, five replicates of each point "lower" limit of quantitation (LOQ), screen cutoff, QC, and upper limit of linearity (ULOL/carryover) were averaged and the %CV was calculated for each analyte. Precision and accuracy were assessed across three days using three replicates of only the screen cutoff and the QC for each analyte. Finally, a comparison of 344 authentic patient positive samples was correlated to the EIA results and confirmation results to determine the false positive and false negative rates as applicable.
Results and Discussion
All analytes seemed to work well with a linear fit through the origin using the single-point calibrator to provide a two-point calibration and benzoylecgonine-D3 as the sole internal standard. Data for the limits of validation can be seen in Table I. The LOQ was 25 ng/mL for every analyte except PCP, which was 4 ng/mL. The MS screen cutoff was set at 100 ng/mL for all analytes except PCP, which was set at 25 ng/mL. All analytes had a concentration of 150 ng/mL in the QC, except PCP, which was at 50 ng/mL. Carryover/ULOL was established at 10,000 ng/mL for amphetamine, benzoylecgonine, MDMA, and methamphetamine; 2500 ng/mL for MDA and MDEA; and 1000 ng/mL for PCP. Because the screen method was concerned more with accuracy in quantitation at the screen cutoff and used a lower calibration point, the carryover/ULOL was just assessed for precision (coefficient of variation [%CV]) and carryover criteria. If the accuracy failed outside of the tolerance, it was deemed acceptable as long as it would not change the positivity of the sample. However, it appeared that even though the single-point calibrator was at a significantly lower concentration than the ULOL, 2–3× the cutoff, the linear range held up to be the same as the original method, >1000 ng/mL depending on the analyte. Requirements for passing included that accuracy of the quantitation be within ±25% and %CV be within ±15%. The passing of the limits of validation indicated that using a two-point calibration with a single IS compound could be successfully used for semiquantitation at least for the analytes in this method.


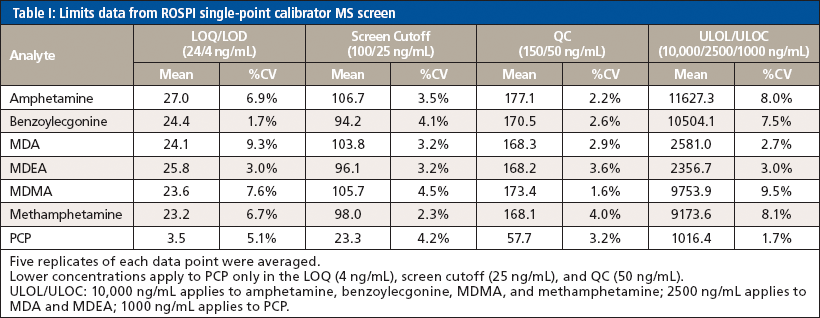
The same acceptance criteria of quantitation within ±25% and %CV within ±15% was still used when assessing the precision and accuracy validation data that can be seen in Table II. However, it was noted that since the QC was a production control that was prepared at a different date and with different working stocks compared to the curve and other calibrators that were also used for quantitation, there was a slightly higher bias with the QC sample. Occasionally, the quantitation accuracy criteria were widened to ±30%, but the %CV criteria were not changed for the precision and accuracy experiment. While this would not have passed the validation to allow use of the method for production, it at least provided a proof-of-concept understanding of the ability to use the two-point calibration with a single IS compound over the course of several days, which was successful based on the precision. Had a new QC solution been prepared from the same stocks, it is reasonable to assume that the accuracy would have passed within the 25% acceptance criteria for all analytes. Furthermore, additional data points (that is, 10 versus 3) would be used to fully validate the method in precision and accuracy before its use in a production environment.


The last proof-of-concept data that were considered were patient comparison data. There were two different assessments of this data. First, a correlation of quantitation between the MS confirmation method and the MS screen method were looked at, which is displayed in Figure 2. Only amphetamine, benzoylecgonine, and methamphetamine had enough patient positives to make the correlation meaningful. Overall, correlation was better than 70% for all three analytes. Benzoylecgonine even had ~98% correlation agreement, which could be because of the fact that benzoylecgonine-D3 was used as the internal standard. Because of the lower correlation with amphetamine and methamphetamine (~71% and ~89%, respectively), it also could be considered if using either the amphetamine-D5 or the methamphetamine-D5 as the IS compound or, in addition to the benzoylecgonine-D3, could improve those correlations, if desired. However, since there tends to be little to no correlation between values from EIA and MS confirmation methods, it could be argued that even ~71% correlation is acceptable.


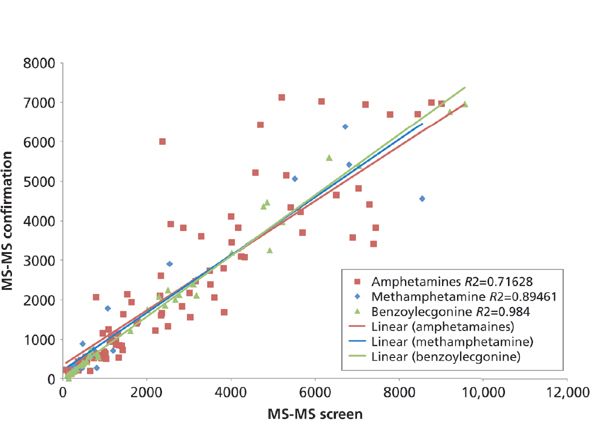
Figure 2: Patient quantitation correlation of MS confirmation and ROSPI MS-screen of amphetamine, benzoylecgonine, and methamphetamine.
Secondly, the patient data from the MS screen were compared to the EIA data to determine the false positive (FPR) and false negative (FNR) rates. It should be noted that all samples were positive biased samples, because they screened positive for at least one of the class compounds by EIA and were then sent for MS confirmation. The samples prepared for the MS confirmation method were then reinjected on the MS screen method to determine whether they screened positive by the MS screen method as well. It should also be noted when assessing the data that the cutoff values used for the EIA and MS screen methods were different in that the MS screen cutoff values are lower than the EIA cutoff values as can be seen in Table III. In general, the MS screen values were lowered to be similar to the MS confirmation reporting cutoff values, with the exception of the PCP cutoff of 25 ng/mL which remained the same between EIA, the MS screen, and MS confirmation. A summary of the data can be seen in Table IV. In general, the MS screen has a lower FPR (%FPR = [number of false positives/total number of positives] × 100). There are a few exceptions. Methamphetamine increases a little from the EIA screen, which could be because the MS screen cutoff was 100 ng/mL and the confirmation cutoff was 125 ng/mL for these samples. Because of the high correlation in quantitation between the two methods, it could be that there is a systematic bias for a higher FPR. Adjusting the cutoff to be 125 ng/mL or higher could help reduce this bias. Interferences, such as phentermine, could also be investigated to determine if that is affecting the FPR for methamphetamine. As for the MDEA and MDMA, while the %FPR was not different, it should be noted that the total number of FPs did decrease significantly from 12 to 1 and the same observation was made for PCP. Benzoylecgonine is the only analyte that actually had a higher incidence of the number of FPs and FPR. It is unclear why this was observed. Unlike methamphetamine, the MS screen and MS confirmation cutoffs are the same, so perhaps there is an interfering compound that could be causing a slight increase in FPs, which would need to be investigated further. Additionally, the FPs for both the EIA and MS screen were compared to determine if the same samples were proving to be problematic by both methods; however, it seemed only about 50% of the FPs were the same between methods, indicating that the reason for a sample being a FP is not the same between methods and that each method has its own flaws and weaknesses.

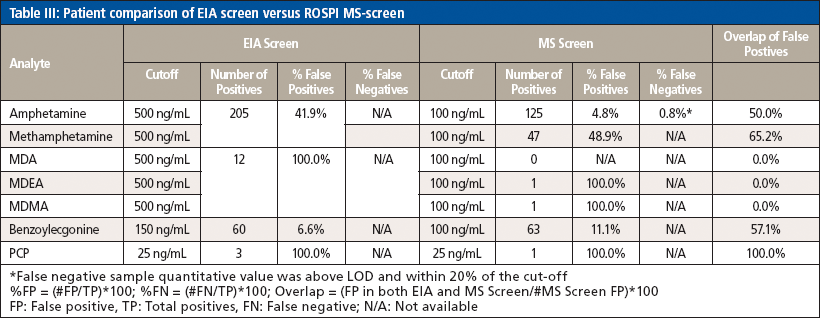
One false negative was observed for amphetamine; this was a case where one sample screened positive by EIA and confirmed positive with the validated MS method, but screened negative by the ROSPI MS method developed here. It should be noted that the quantitative value determined by the single-point calibration was within 20% of the cutoff value, so perhaps using a different IS compound could improve the accuracy and correlation between patient samples to reduce the change of FNs. However, the FNR (%FN = [false negative/total positives] × 100) is still low enough to be considered acceptable. The FNR could not be calculated for EIA in this validation because any sample that screens negative by EIA is not processed by the MS confirmation method.
Conclusion
Overall, the data show that ROSPI is a viable MS screening alternative to EIA and could provide cost savings and improvements in FPR. Further validation experiments should be considered before using this method in a production environment. In particular, to be more time efficient, using a guard cartridge that is shorter (for example, 4 mm) and an autosampler alternative could be investigated to improve the cycle time further. Many autosamplers now have a much shorter cycle time, ~10–20 s, compared to the one used in our study, which can only be shortened to ~0.9 min. However, what was established here still met the study's goals and reduced the time from the confirmation method by ~50%. Increasing the flow rate or investigating duplexing and multiplexing could also provide more time savings. As for additional validation experiments, a more extensive precision and accuracy experiment with more concentrations and replicates along with an interference study appears to be the most crucial. However, matrix effect and signal suppression experiments should be considered as well, especially if a shorter cartridge is used that increases the coelution of interferences and signal suppressing compounds, which was observed in high-throughput MS (6), the logical endpoint of shortening the column.
The time required for completion of a single screen (that is, one sample) was dictated by the length of the guard column inasmuch as the flow rate was not changed. While the cycle time observed herein met the criteria for an effective screen, faster times have been observed in work using guard "filters" as short as 4 mm. This experimental work has also shown that the optimal combination of "column" length and flow rate remain empirical with issues such as tailing and peak shape requiring specific combinations of these parameters. The smaller "columns" used herein are less expensive than conventional analytical columns and seem to hold up well to the dilute and shoot paradigm used in this work. Sample preparation (that is, solid-phase extraction or supported liquid extraction) can be used, but it would increase the cost per sample markedly. Lastly, the sample can be prepared in vials or 96-well plates of conventional format to further simplify the process and minimize the expense.
This ROSPI proof of concept also showed that using a two-point calibration curve with a singular IS compound can afford sufficient semiquantitation used in a screening setting to determine the positivity of samples. While these compounds were successful in maintaining the original MS confirmation method ULOL and carryover limit, there may be some compounds that do not have the same behavior, but within a reasonable range around the cutoff value quantitation can be achieved with the same accuracy as an MS confirmation method. It was also demonstrated that while EIA values are known to have little to no correlation to MS quantitative methods, it can be seen here that MS screen methods have high correlation between the MS screen value and the MS quantitative value. This could have an impact on the cutoff values used for screening relative to the MS confirmation cutoff. Typically, EIA screen cutoffs are higher than MS confirmation methods; however, the correlation data indicates that MS screen cutoff should be lower than EIA screen cutoffs and more similar to the MS confirmation cutoff. For the compounds that have lower positivity rates, MDA, MDEA, MDMA, and PCP that were not correlated, a benefit for these compounds is the overall reduction in the number of positives and that now the cost of these screens is included with the higher volume-positive screens of amphetamine, methamphetamine, and benzoylecgonine. This is beneficial in cost-savings since there is a limited shelf-live for the screening solutions that could go bad with these lower demanding screens. With an MS-based screen there is not as much concern for the expiration of standards, especially since they can be used for the other compounds as well. Overall, with proper method development and validation ROSPI-MS screening is a reasonable time- and cost-efficient alternative to EIA screening.
Erin C. Strickland, Ian Shapiro, and Gregory L. McIntire are with Ameritox, Ltd. in Greensboro, North Carolina. Direct correspondence to: Greg.mcintire@ameritox.com
References
(1) C.C. Ozbal, W.A. LaMarr, J.R. Linton, D.F. Green, A. Katz, T.B. Morrison, and C.J.H. Brenan, Assay Drug Dev. Technol.2, 373–382 (2004).
(2) A.K. Quercia, W.A. LaMarr, J. Myung, C.C. Ozbal, J.A. Landro, and K.J. Lumb, J. Biomol. Screen.12, 473–480 (2007).
(3) C.D. Forbes, J.G. Toth, C.C. Ozbal, W.A. LaMarr, J.A. Pendelton, S. Rocks, R.W. Gedrich, D.G. Osterman, J.A. Landro, and K.J. Lumb, J. Biomol. Screen.12, 628–634 (2007).
(4) K.B. Lim, C.C. Ozbal, and D.B. Kassel, J. Biomol. Screen.15, 447–452 (2010).
(5) M.K. Highkin, M.P. Yates, O.V. Nemirovskiy, W.A. LaMarr, G.E. Munie, J.W. Rains, J.L. Masferrer, and M.M. Nagiec, J. Biomol. Screen.16, 272–277 (2011).
(6) W. Jian, M.V. Romm, R.W. Edom, V.P. Miller, W.A. LaMarr, and N. Weng, Anal. Chem.83, 8259–8266 (2011).
(7) M. Razavi, L.E. Frick, W.A. LaMarr, M.E. Pope, C.A. Miller, N.L. Anderson, and T.W. Pearson, J. Proteome Res.11, 5642–5649 (2012).
(8) S.E. Hutchinson, M.V. Leveridge, M.L. Heathcote, P. Francis, L. Williams, M. Gee, J. Munoz-Muriedas, B. Leavens, A. Shillings, E. Jones, P. Homes, S. Baddele, C. Chung, A. Bridges, and A. Argyrou, J. Biomol. Screen.17, 39–48 (2012).
(9) X. Wu, J. Wang, L. Tan, J. Bui, E. Gjerstad, K. McMillan, and W. Zhang, J. Biomol. Screen.17, 761–772 (2012).
(10) N.R. Parikh, M. Romm, and V.P. Miller, Spectrosc. Eur.25, 15–17 (2013).
(11) J. Wu, C.S. Hughes, P. Picard, S. Letarte, M. Gaudreault, J. Levesque, D.A. Nicoll-Griffith, and K.P. Bateman, Anal. Chem. 79, 4657–4665 (2007).
(12) P.B. Fayad, M. Prevost, and S. Sauve, Anal. Chem.82, 639–645 (2010).
(13) J.G. Swales, R.T. Gallagher, M. Denn, and R.M. Peter, J. Pharm. Biomed. Anal.55, 544–551 (2011).
(14) L. Viglino, M. Prevost, and S. Sauve, J. Environ. Monit.13, 583–590 (2011).
(15) O. Heudi, S. Barteau, P. Picard, P. Tremblay, F. Picard, and O. Kretz, J. Pharm. Biomed. Anal.54, 1088–1095 (2011).
(16) D.P. Mohapatra, S.K. Brar, R.D. Tyagi, P. Picard, and R.Y. Surampalli, Talanta99, 247–255 (2012).
(17) K. Badjagbo and S. Sauve, Anal. Chem.84, 5731–5736 (2012).
(18) I. Beattie, A. Smith, D.J. Weston, P. White, S. Szwandt, and L. Sealey, J. Pharm. Biomed. Anal. 59, 18–28 (2012).
(19) P. Lemoine, A. Roy-Lachapelle, M. Prevost, P. Tremblay, M. Solliec, and S. Sauve, Toxicon.61, 165–174 (2013).
(20) F.M. Fernandez, R.B. Cody, M.D. Green, C.Y. Hampton, R. McGready, S. Sengaloundeth, N.J. White, and P.N. Newton, ChemMedChem1, 702–705 (2006).
(21) T.M. Vail, P.R. Jones, and O.D. Sparkman, J. Anal. Toxicol.31, 304–312 (2007).
(22) Y. Zhao, M. Lam, D. Wu, and R. Mak, Rapid Commun. Mass Spectrom.22, 3217–3224 (2008).
(23) E. Jagerdeo and M. Abdel-Rehim, J. Am. Soc. Mass Spectrom. 20, 891–899 (2009).
(24) L.K. Ackerman, G.O. Noonan, and T.H. Begley, Food Additives & Contaminants: Part A26, 1611–1618 (2009).
(25) L. Vaclavik, T. Cajka, V. Hrbek, and J. Hajslova, Anal. Chim. Acta645, 56–63 (2009).
(26) A.J. Dane and R.B. Cody, Analyst135, 696–699 (2010).
(27) E.S. Chernetsova, P.O. Bochkov, M.V. Ovcharov, S.S. Zhokhov, and R.A. Abramovich, Drug Test. Anal.2, 292–294 (2010).
(28) J.M. Nilles, T.R. Connell, S.T. Stokes, and H.D. Durst, Propellants, Explos., Pyrotech.35, 446–451 (2010).
(29) M. Zhou, J.F. McDonald, and F.M. Fernandez, J. Am. Soc. Mass Spectrom.21, 68–75 (2010).
(30) E.S. Chernetsova, G.E. Morlock, and I.A. Revelsky, Russ. Chem. Rev.80, 235 (2011).
(31) T. Cajka, K. Riddellova, M. Tomaniova, and J. Hajslova, Metabolomics7, 500–508 (2011).
(32) W.C. Samms, Y.J. Jiang, M.D. Dixon, S.S. Houck, and A. Mozayani, J. Forensic Sci.56, 993–998 (2011).
(33) Z. Takats, J.M. Wiseman, B. Gologan, and R.G. Cooks, Science306, 471–473 (2004).
(34) Z. Takats, J.M. Wiseman, B. Gologan, and R.G. Cooks, Science306, 471–473 (2004).
(35) H. Chen, N.N. Talaty, Z. Takats, and R.G. Cooks, Anal. Chem.77, 6915–6927 (2005).
(36) J. Shiea, M. Huang, H. Hsu, C. Lee, C. Yuan, I. Beech, and J. Sunner, Rapid Commun. Mass Spectrom.19, 3701–3704 (2005).
(37) J.P. Williams and J.H. Scrivens, Rapid Commun. Mass Spectrom.19, 3643–3650 (2005).
(38) Z. Takats, I. Cotte-Rodriguez, N. Talaty, H. Chen, and R.G. Cooks, Chem. Commun.15, 1950–1952 (2005).
(39) Z. Takats, J.M. Wiseman, and R.G. Cooks, J. Mass Spectrom.40, 1261–1275 (2005).
(40) I. Cotte-Rodriguez and R.G. Cooks, Chem. Commun.28, 2968–2970 (2006).
(41) S.E. Rodriguez-Cruz, Rapid Commun. Mass Spectrom.20, 53–60 (2006).
(42) T.J. Kauppila, J.M. Wiseman, R.A. Ketola, T. Kotiaho, R.G. Cooks, and R. Kostiainen, Rapid Commun. Mass Spectrom.20, 387–392 (2006).
(43) Z. Miao and H. Chen, J. Am. Soc. Mass Spectrom. 20, 10–19 (2009).
(44) J.M. Wiseman, C.A. Evans, C.L. Bowen, and J.H. Kennedy, Analyst135, 720–725 (2010).













The State of Forensic Science: Previewing an Upcoming AAFS Video Series
March 10th 2025Here, we provide a preview of our upcoming multi-day video series that will focus on recapping the American Academy of Forensic Sciences Conference, as well as documenting the current state of the forensic science industry.

Rapid, Portable Mid-Infrared Spectroscopy Identifies Aflatoxins in Peanuts
March 10th 2025Researchers have developed a portable mid-infrared (IR) spectroscopic method combined with chemometric analysis to rapidly and non-destructively detect aflatoxin contamination in Aspergillus-infected peanuts. This approach offers a field-deployable alternative to traditional wet chemistry methods, with high sensitivity and specificity in identifying toxic metabolites such as aflatoxins.



Previewing the American Academy of Forensic Sciences Conference
February 14th 2025This year, the American Academy of Forensic Sciences Conference is taking place from February 17–22, 2025. We highlight the importance of spectroscopy in this field and why we’re covering the conference this year.

