Do You Really Understand the Cost of Noncompliance?
Two recent warning letters show that the US FDA is substantially increasing the amount of remediation work it requires for companies to correct data integrity noncompliance. That work can be very expensive—far exceeding the cost of ensuring compliance in the first place.
English is hopeless as there is no word for it. Spanish? Nope. French? Forget it. Chinese? Not a clue. In contrast, German has the word for it. What is it? Schadenfreude. Huh? Schadenfreude is defined by Wikipedia as the experience of pleasure, joy, or self-satisfaction that comes from learning of or witnessing the troubles, failures, or humiliation of another.Has this column turned into a language lesson? No. Rather, my goal is to teach you that it is better to be compliant than not.
Previously, my focus in previous “Focus on Quality” columns has been mainly on instances of noncompliance found in FDA Form 483 observations and warning letters, such as a recent review of cases of infrared (IR) spectrometer noncompliance (1). However, in this installment, I discuss the extensive and expensive remediation activities required by the FDA in two July 2020 warning letters issued for data integrity violations by two U.S. companies, Stason Pharmaceuticals and Tender Corporation (2,3).
What is the relationship of this to schadenfreude, you may ask? Wait until you see the scope and detail of the work required by the FDA to remediate the noncompliance by these two companies. It looks like the FDA is losing patience with the pharmaceutical industry and is sending an unmistakable message to companies to get their data integrity act together.
Use schadenfreude as described here to serve as a warning to check how compliant your laboratory processes and systems are now. Being inspection-ready is always preferable to having an inspector uncover noncompliant bodies that have been buried. Before we get into the miasma of these noncompliances, we need first to understand and discuss the costs of compliance and noncompliance.
Is That All a Regulation Says?
The problem in the pharmaceutical industry is that many analytical chemists in the industry have never read the regulations they have to comply with, because the regulations have been interpreted in the past by the organization. However, reading, understanding, and interpreting regulations is critical. Take for example the U.S. Good Manufacturing Practices (GMP) regulations for equipment in 21 CFR 211.63. We can boil down the 35 words of this clause into three simple requirements, that instruments:
- have appropriate design
- have adequate size
- be suitably located for intended use (4).
That’s all it says. However, these words need to be interpreted in combination with USP <1058> (5):
- A specification for an analytical instrument and any software must be documented in a user requirements specification (URS). The URS should contain statements about adequate size (for example, capacity, such as maximum sample numbers in a run) of the instrument or number of concurrent users if a networked system.
- Design qualification (DQ) is required to demonstrate that the right instrument or system has been selected.
- The instrument and any associated software must be installed correctly (installation qualification).
- The instrument and any software must undergo an operational qualification and user acceptance testing against the URS to confirm fitness for intended use. This phase can include an appropriate mixture of calibrating, qualifying, and testing the whole system while establishing a baseline against which operation of the instrument can be monitored.
- Monitoring the operation of the instrument over time against the baseline and user requirements (performance qualification) is required.
Given that the regulations state only the minimum required elements, these regulations have spawned an industry of writers to interpret these regulations through regulatory guidance documents, a pharmacopeial general chapter, publications from scientific societies, and articles by individuals like me (5–10).
Even with all these publications, there is still the choice of how to implement these regulations in an individual laboratory for each analytical instrument and computerized system. Interpretation of pharmaceutical regulations is always a balance between the cost of compliance against the cost of noncompliance. Remember also that any work in a GMP-regulated laboratory must be scientifically sound under 21 CFR 211.160(b) (4). Figure 1 illustrates the balance between the costs of compliance and noncompliance.


Figure 1: Understanding the balance between the costs of compliance and noncompliance (10,13).
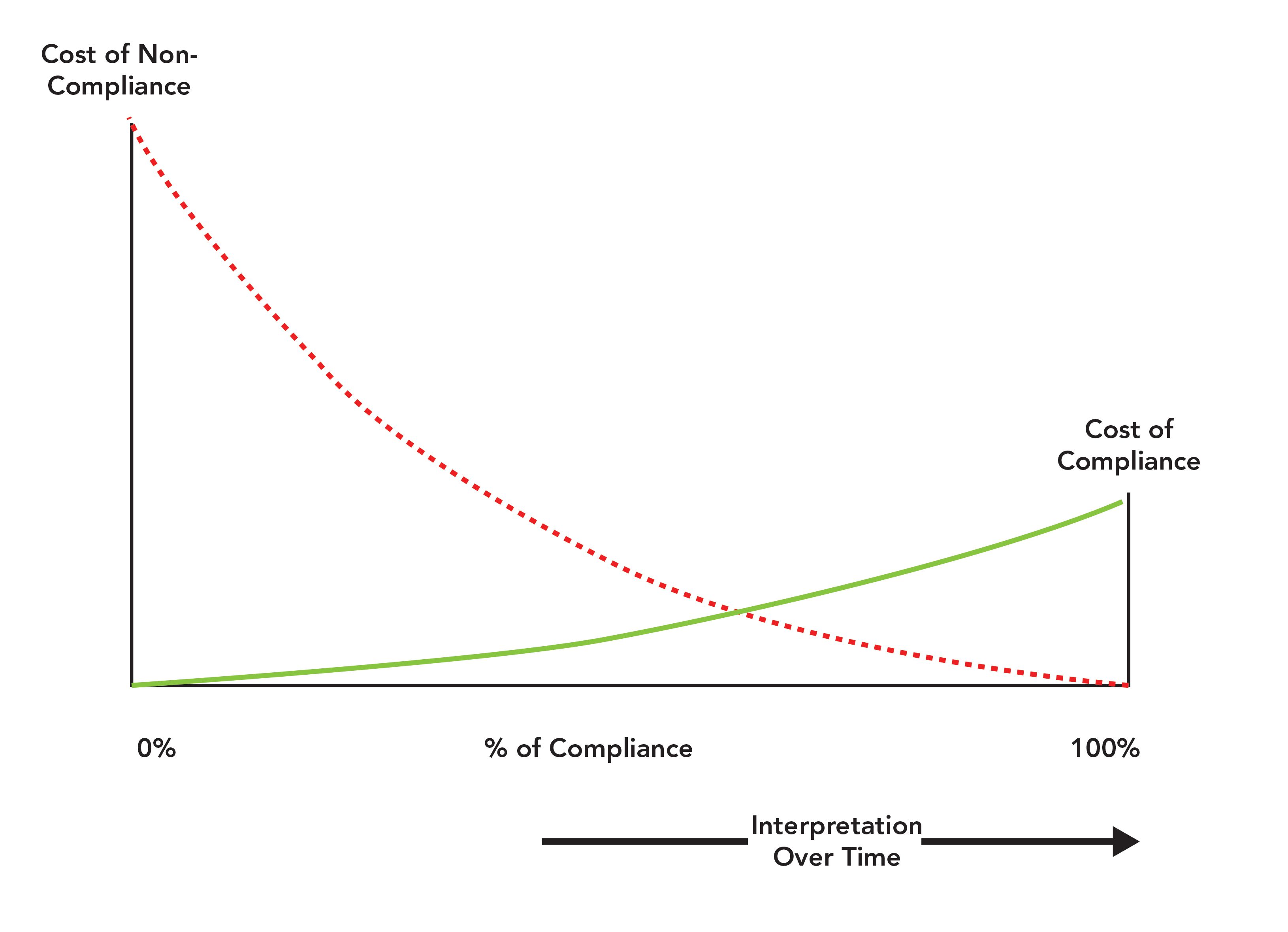
Cost of Compliance vs. Cost of Noncompliance
Risk management is one of the requirements for the pharmaceutical industry following the publication of the FDA’s GMPs for the 21st Century document and the ICH Q9 guideline on quality risk management (11,12). The work required in a regulated laboratory is dependent on a justified and documented risk assessment that must also be scientifically sound. This discussion can be summarized as the balance between the cost of compliance and the cost of noncompliance. Each laboratory makes decisions along a spectrum that ranges from doing nothing to doing everything possible, and this decision determines how much regulatory and business risk a company wishes to mitigate or carry as well as how much money the company wishes to spend.
The left-hand vertical axis of Figure 1 is the cost of noncompliance and the right-hand vertical axis shows the cost of compliance. You will note that the cost of noncompliance axis is much bigger than the cost of compliance axis. One viewpoint is that one axis is logarithmic, and the other is linear. Guess which one is linear? This is one of the balances you need to consider: The right-hand side shows the cost of doing it right the first time, and the left-hand side is the cost of getting caught. Fixing a regulatory problem that has been identified in a warning letter is always more expensive than doing the right thing the first time, or even of finding a problem and fixing it yourself. If anyone is in doubt about the cost of noncompliance for data integrity violations, I suggest that you read a consent decree, such as that for Ranbaxy (14). In that consent decree, the cost of noncompliance can be quantified as hundreds of millions of dollars.
In Figure 1, the horizontal axis is the percentage of compliance from 0 to 100%. The only fixed points are at the ends of the scale where 0% indicates no control of the process or system and 100% is where anything that can be compliant is compliant. In between 0 and 100% is a relative scale of compliance. The major point to note is that this scale is not fixed but moves as indicated by the arrow at the bottom of the figure. However, the direction of movement is only one way and that is to the right! To understand this point, consider the situation with data integrity. The FDA GMP regulations have changed little since 1978 and have always contained data integrity requirements. However, since the Able Laboratories fraud case in 2005 and the discovery of industrial-scale data falsification and poor data management practices, we have seen regulatory authorities issue guidance documents on the topic and enforce the regulations more strictly (15–20). Because of the proactive monitoring by regulatory authorities, the compliance scale has moved to the right.
Compliance Awareness?
Because data integrity has been a major compliance topic for the past 15 years, you would think that organizations would have taken the hint and started assessment and remediation projects already. Apparently, this is not the case. There have been many past warning letters highlighting the issues that are common in the two warning letters that we review in this column. Notwithstanding the availability of those letters on the FDA’s website, apparently little if anything was been done at Stason Pharmaceuticals or Tender Corporation to even assess if there were any problems; such an effort was solely the responsibility of senior management. Also, little if any effort had been made at these companies to assess the compliance landscape, see the trends in inspections, and plan ahead. Then again, even if there was the knowledge of compliance trends at those companies, would either organization have done anything different? One has to wonder. For those working in organizations that are aware of the data integrity issues and are working to remediate the problems, you can have a little schadenfreude smirk.
Who is on the Naughty Step Today?
We are now ready to consider the cost of noncompliance of two warning letter remediation plans mandated by the FDA (2,3). These are as follows:
- Stason Pharmaceuticals Inc. received two citations against 21 CFR 211.192 and 21 CFR 211.68(b). There were major problems with out of specification (OOS) investigations that resulted in a major list of remediation tasks. Our interest is in the second citation for failure to exercise appropriate controls over computerized systems so that only authorized individuals can make changes to records.
- Tender Corporation has three citations, under 21 CFR 211.194(a), 21 CFR 211.160(b), and 21 CFR 211.192. Here the citations were for failure to have complete data available from testing by deleting data, although there is also mention of common login and password for access, which is a 211.168(b) requirement. There was also a failure to have scientifically sound test methods and assessment of method problems. The final citation was for a failure to have a procedure for compliant handling and inadequate complaint investigations.
The main citations from both warning letters are shown in Figure 2. Interestingly, both organizations are U.S. companies and not located in India or China, reinforcing that data integrity is a global problem. Both warning letters were issued by the FDA’s Division of Pharmaceutical Quality Operations.
What is interesting is that Stason is cited under 21 CFR 211.68(b) for failing to control computer systems and Tender is cited under 21 CFR 211.160(b) for a failure of laboratory controls. However, each company is required to submit a comprehensive remediation plan for data integrity but from different clauses of the U.S. GMP regulations. So what? The what is a nearly identical list of remediation activities contained in both FDA warning letters that support my contention that the cost of non-compliance is significantly larger than the cost of compliance.
A Comprehensive Remediation Plan
Both warning letters require comprehensive corrective action and preventive action (CAPA) plans, shown at the bottom of Figure 2, and the wording is identical:


Figure 2: Overview of the Stason Pharmaceuticals and Tender Corporation warning letters.

A comprehensive, independent assessment and corrective action and preventive action (CAPA) plan for computer system security and integrity. Include a report that identifies vulnerabilities in the design and controls. Also include appropriate remediations for each of your laboratory computer systems. This should include but not be limited to…
Let us analyze this single paragraph in some detail:
- Comprehensive: An extremely detailed and extensive report is required. Some of the words and phrases used to describe the remedial action required include describe in detail, strict, on-going control, enhanced, and comprehensive review. This plan is not going to be written on the back of an envelope. Nor will it be cheap.
- Independent assessment: The work must be led and conducted by an external consulting company with extensive knowledge of data integrity, regulations, and computerized systems. It will also require much input from laboratory staff, which will slow down analytical work while trying to assess and remediate processes, systems, and documentation. A consulting opportunity.
- Computer security and integrity:Each computerized system in the laboratory needs to be assessed from the perspectives of unique user identities, password use, user roles with associated access privileges that avoid conflicts of interest, and application configuration to ensure protection of electronic records.
- Vulnerabilities in design and controls: Data process mapping needs to be conducted on all systems. For each workflow in a system the records generated need to be identified, and the controls in place (assuming that there are some) and the data vulnerabilities need to be noted. Data process mapping is an iterative process involving a facilitator and subject-matter experts from the laboratory and, where appropriate, the IT group. It does not state how many instruments or computerized systems are in either laboratory, but for a medium-sized laboratory, there might be 10–25 instruments.
- Appropriate remediations: The list cites both procedural controls and training for staff but also mentions technical controls (and the associated validation costs) for long-term resolution of the data integrity problems.
At the end of every warning letter is the following paragraph:
The violations cited in this letter are not intended to be an all-inclusive list of violations that exist at your facility. Inspections and audits sample and only identify problems seen during the visit.
Inspections and audits sample and only identify problems seen during the visit. The regulatory expectation is that a systems approach is taken and if there is a problem with user account management seen with one computerized system, then an assessment of all systems should be undertaken. In addition, during the assessment of processes and systems a new non-compliance is identified, fix it! This is in contrast to the traditional QA mentality to just answer the question asked and not to think wider.
From a single paragraph in each warning letter you can sense that this remediation plan will be extensive and expensive. This program of work is not a two-minute job and with external involvement in all aspects of the preparation, this work will not be cheap. Remember the sentence at the end of the paragraph: “This should include and not be limited to…more work may be required than listed in the warning letter.” This is a consulting opportunity by invitation of the FDA. My spectroscopic schadenfreude-o-meter is starting its inexorable rise into the red zone and warning lights are flashing down at quality control!
It is interesting that many organizations claim that they never have any money available for improvements or time to do compliance work. It is strange that following the receipt of a 483 and especially a warning letter, money and resources flow like water over Niagara Falls. Now you can begin to see how the cost of non-compliance is much more than the cost of compliance. With the former, you have the boot of a regulatory agency inserted in places it should not be inserted, combined with an urgent timetable from the company to resolve the non-compliance. In comparison, a well documented risk management approach that is defendable has a fraction of the cost. Not convinced yet that the cost of compliance is less than the cost of noncompliance? Before we see the evidence to support my contention, as we enter into the valley of noncompliance death, we need to discuss my data integrity model. This is necessary to understand the scope of data integrity within the context of a pharmaceutical quality system.
Reprising A Data Integrity Model
The analytical portion of my data integrity model has featured in this column before and the full model in other publications of mine (10,13,21–23). It consists of four layers analogous to building a house plus quality oversight and is presented in Figure 3. This model is important because we map the citations in the warning letters against the four levels of the model. The analogy with building a house is that if there is no foundation, the house collapses. This analogy is important to remember when we review the warning letters and analyze the remediation. Although the full model shows three levels of production and quality control (QC) analysis, we will only consider the foundation, analytical levels, and quality assurance (QA) oversight in the analysis of the warning letters. The layers of the model are:
- Foundation (F): Data governance, which includes management leadership, quality culture, ethos, and training for data integrity including good documentation practices.
- Level 1 (L1): The right analytical instrument and computerized system qualified and validated for intended use.
- Level 2 (L2): The right analytical procedure for verification or validation under actual conditions of use.
- Level 3 (L3): Executing the validated procedure using qualified instruments and validated computerized systems with staff trained in data integrity operating in an open culture.
- Quality oversight (QA): Checks, audits, and investigations to ensure work at all levels has been performed correctly.


Figure 3: A data integrity model (taken from reference [13]).
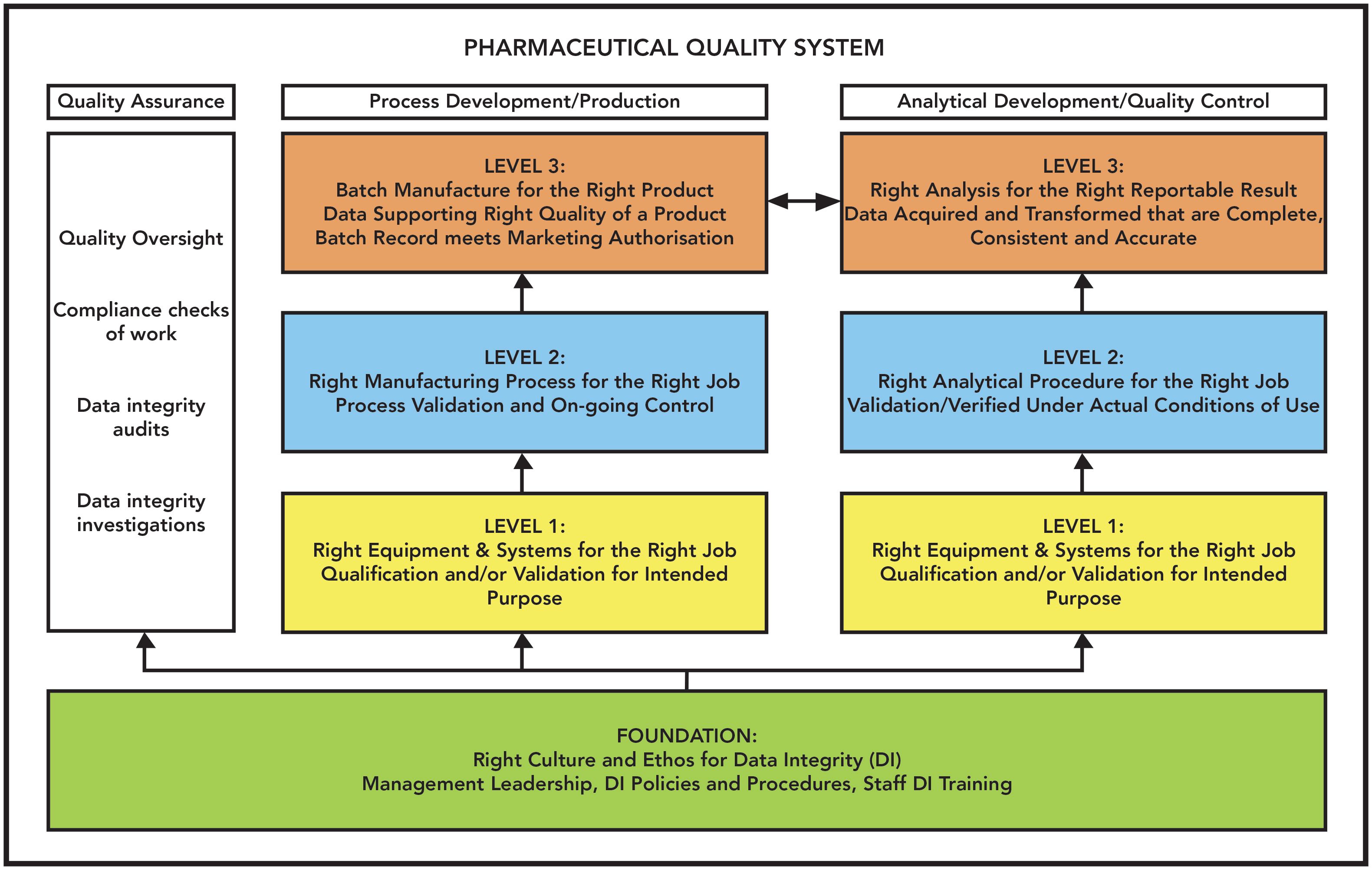
The whole of this model must exist within a company’s pharmaceutical quality system. Each part of the FDA-required remediation plan will be mapped to one or more parts of this model using the annotations in parenthesis in the bullet list above.
Getting into the Detail
Here’s where we go into detail for the remediation CAPA plan. If you think the overview discussed above was bad, wait until you see what is coming. Figure 3 shows the minimum elements of the data integrity remediation required by the FDA of Stason Pharmaceutical and Tender Corporation. Item 12 in Figure 3 is exclusive to the latter company. Visualizing each plan is essential to have a better understanding of the scope and detail of the remediation required, which are extensive. Please note that the elements of the plan in Figure 3 have had the text edited so that the figure is manageable.
In contrast, Table I presents the data integrity remediation tasks in both the Stason Pharmaceuticals and Tender Corporation warning letters, and the one additional requirement required of Tender Corporation is added at the bottom of this table. Table I consists of three columns:
- The left-hand column lists the verbatim requirement copied from the
warning letters. - The middle column shows the requirement mapped against the data
integrity model. - The right-hand column shows my comments on the remediation requirements



Remediation Requirements Review
We discussed the main features of a data integrity model earlier and each of the remediation requirements of the FDA in Table I are mapped against the layers of this model. Figure 4 shows the required remediation activities mapped visually against the model. This mapping is illuminating because both organizations have failed to implement basic GMP requirements in terms of data governance and associated training at the foundation and failed totally at level 1 to ensure that analytical instruments and computerized systems are fit for their intended use.


Figure 4: Visualization of the Stason Pharmaceuticals and Tender Corporation remediation plans.
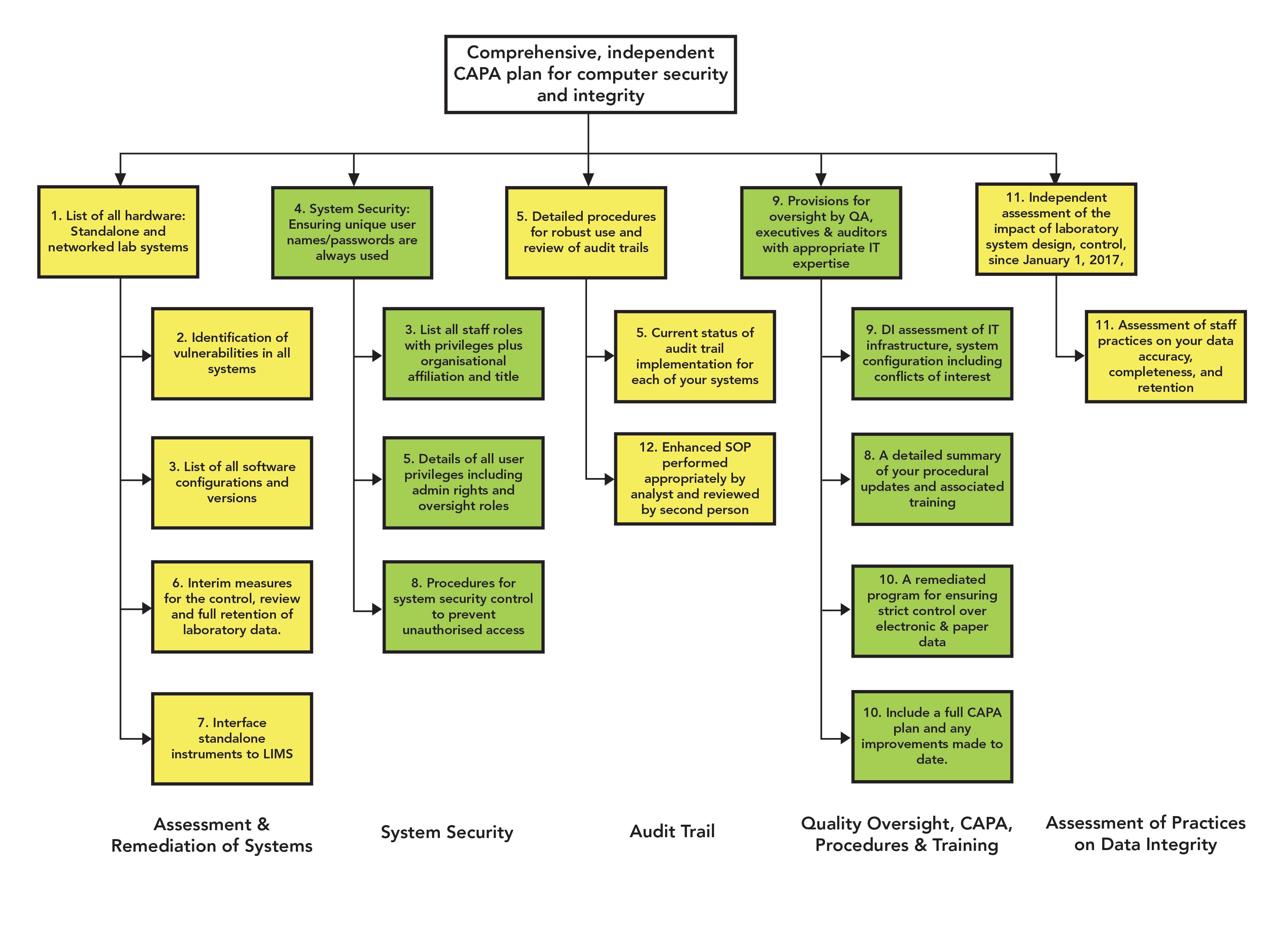
This figure also reinforces a key point that has been made since the inception of this model: If you don’t get the lower levels of the data integrity model correct, then whatever data integrity efforts are implemented at the upper layers become worthless. Of interest is the fact that level 2 is not mentioned at all in the remediation requirements because of the scope and depth of the non-compliances cited in Table I were such a shambles that the inspectors did not need to look closely at how analytical procedures were verified or validated.
The issues mapped to level 3 are, in part, a direct result of failures at the foundation and level 1. For example, because audit trails have not been implemented and validated at level 1, there is a consequential requirement for a procedure and training on how to review the entries during batch analysis at level 3. Security and access control appear conspicuous by their absence before the inspection as there is a major remediation effort to attribute work to individuals and avoid conflicts of interest when accessing systems, another level 1 failure. The failure to meet the requirement for complete data to be generated during testing is a fundamental failure in training staff in good documentation practices and data integrity including the meaning of complete data at the foundation level.
Continuing with omissions in the foundation, the spotlight now turns full beam on management. Management is not treated very kindly as there appears to be an abject failure of executive leadership with respect to data integrity and data governance. This is a fully deserved citation as data integrity begins and ends with executive management. Management is responsible for the pharmaceutical quality system and this is made crystal clear in GMP and good laboratory practices (GLP) regulations (24–26) as well as in regulatory guidance documents on data integrity (16–20). Any data integrity program requires resources, time, and funding for the assessment and remediation of processes and systems including updating or replacing inadequate systems. Executive management bears sole responsibility for this work.
The QA departments in these firms also appear to be inept, given that FDA’s remediation has the explicit requirement for quality staff with IT knowledge. It seems likely that the focus of quality oversight was only on paper records, possibly with paper printouts from the computerized systems being defined as the “raw data.” This is despite the fact that the FDA has had a level 2 guidance on why electronic records take priority over paper printouts available since 2010, which is publicly available on the agency’s web site (27). This position was reiterated in the 2018 data integrity guidance from the agency (19).
There is an initial remediation emphasis on procedural controls but the longer-term focus is directed by the FDA to technical controls. Overall, there should be elimination of paper by interfacing instruments, such as pH meters, and analytical balances to a laboratory information management system (LIMS). This is a crucial approach as procedural controls are operated by humans but consistency and identifying errors are not a given. Technical controls that have been validated can be operated consistently and enforce a procedure and hence GXP compliance, this is a far better approach compared to procedural controls.
Sometimes I get a feeling that an independent assessment of the current working practices will open one or more cans of worms for the company, and my schadenfreude-o-meter is ready to tick up a notch or two.
The focus of the FDA’s remediation requirements at level 1 of the model appears to indicate that both organizations did little more than install all analytical systems and then operate them in default mode. The systems themselves were either poorly designed (no database or audit trails were never turned on) or badly implemented, because it was possible to delete data. Because there are numerous citations for data deletions or failing to report all data, the remediations will be very expensive, coupled with the two independent assessments of systems and working practices.
A Bonus Remediation Task
In addition to the work required under the comprehensive and independent CAPA plan listed in Table I, there is a second major data integrity remediation requirement for Stason:
A complete assessment of documentation systems used throughout your manufacturing and laboratory operations to determine where documentation practices are insufficient. Include a detailed CAPA plan that comprehensively remediates your firm’s documentation practices to ensure you retain attributable, legible, complete, original, accurate, contemporaneous records throughout your operation (2).
How best to summarize this paragraph? Your quality management system is terrible. Fix it. This is a massive project in addition to all the other remediation work. Here the whole organization’s quality management system (QMS) and all methods of recording regulated activities, both in the laboratory and in production, have been brought into question. If the agency cannot trust the data, what hope is there for the company other than a complete reassessment of policies and procedures within the QMS? This work must run in parallel with the data integrity remediation discussed above. There will be multiple interactions between the two projects to ensure attributable, legible, contemporaneous, original, and accurate (ALCOA) principles are applied while completing data and record integration into the foundation of what the company does. This work applies not just to paper but also to hybrid and electronic records. This extensive remediation project can be mapped to the foundation level of the data integrity model.
Noncompliance Can Seriously Damage Your Wealth
When a company receives an FDA warning letter, there are several consequences, many of which will result in additional costs that we have not covered yet.
- Reactive Compliance Approach: Laboratories that have a reactive compliance approach, summarized as “wait until a noncompliance is found and then fix it,” will find that the exponential cost of remediation action makes this approach extremely expensive, dangerous and foolish as seen in Figure 1. A proactive compliance approach of “do it right first time” is the only way to work now.
- Regulatory Credibility: A laboratory’s regulatory credibility is lost, not just with the FDA, but with global regulators as inspection information is shared under mutual recognition agreements. For example, when FDA places a foreign company on import alert, Health Canada frequently follows suit even if they have not been involved with the inspection. The result will be increased scrutiny during subsequent inspections from all authorities. This means ensuring that the organization is in a state of constant inspection readiness. For serial offenders, there is an option of a consent decree of permanent injunction that binds the company to be compliant with the regulations in perpetuity and that also may be associated with large fines (14).
- Informing Your Sponsors: This entire situation is even worse for a contract research organization (CRO) or contract manufacturing organization (CMO). Here one lucky individual draws the short straw to phone all sponsors to inform them of their regulatory failings. This task is guaranteed to raise any individual’s blood pressure and may lead to a loss of business with existing and potential sponsors. In some cases, work supporting a regulated product may need to be repeated, as sponsors found with studies carried out by Cetero Research (28,29).
- Share Price Impact: Information about a warning letter or even the possibility of one can result in a fall in the company share price. For example, on February 15, 2019, the price of shares of Dr. Reddy’s Laboratories fell nearly 29% in a morning on the Bombay Stock Exchange before ending the day more than 4% lower than its previous close. This was because of the fear of a further FDA warning letter for the company. In a situation like this, management needs to think about whether their stock options are worth anything.


Figure 5: Mapping the Stason Pharmaceuticals and Tender Corporation warning letter citations to the data integrity model.

Summary
We have discussed how to interpret pharmaceutical regulations with the careful balance between the cost of compliance with the cost of non-compliance. The two warning letters discussed here clearly demonstrate that it is easier and cheaper to be compliant rather than face the consequences of remediating noncompliance. Will organizations listen, eliminate paper, and move to automated processes with validated technical controls to ensure compliance and data integrity? That is the $64,000 question (cost of compliance) or $64,000,000 question (cost of non-compliance). If your organization is compliant, you can experience schadenfreude.
Acknowledgments
I would like to thank John English whose work of posting FDA 483 observations and warning letters on LinkedIn led to the genesis of this article, and Chris Burgess, Paul Smith, and Kevin Roberson for helpful comments during preparation.
References
- FDA, Warning Letter Stason Pharmaceuticals, Inc. (FDA, Silver Spring, Maryland, 2020).
- FDA, Warning Letter Tender Corporation (FDA, Silver Spring, Maryland, 2020).
- P.A. Smith and R.D. McDowall, Spectroscopy 34(9), 22–28 (2019).
- “Current Good Manufacturing Practice for Finished Pharmaceutical Products,“ in 21 CFR 211 (Food and Drug Administration, Sliver Spring, Maryland, 2008).
- General Chapter <1058> “Analytical Instrument Qualification,” in United States Pharmacopoeia (United States Pharmacopeial Convention, Rockville, Maryland, 2002).
- FDA, Guidance for Industry General Principles of Software Validation (FDA, Rockville, Maryland, 2002).
- ISPE, Good Automated Manufacturing Practice (GAMP) Guide (International Society for Pharmaceutical Engineering, Tampa, Florida, 2nd ed., 2012).
- ISPE, Good Automated Manufacturing Practice (GAMP) Guide version 5 (International Society for Pharmaceutical Engineering, Tampa, Florida, 2008).
- R.D. McDowall, Spectroscopy 32(9), 24–30 (2017).
- R.D.McDowall, Validation of Chromatography Data Systems: Ensuring Data Integrity, Meeting Business and Regulatory Requirements (Royal Society of Chemistry, Cambridge, United Kingdom, 2nd ed., 2017).
- FDA, Pharmaceutical cGMPs for the 21st Century: A Risk-Based Approach (FDA, Rockville, Maryland, 2002).
- International Conference on Harmonization, ICH Q9, Quality Risk Management (ICH, Geneva, Switzerland, 2005).
- R.D.McDowall, Data Integrity and Data Governance: Practical Implementation in Regulated Laboratories (Royal Society of Chemistry, Cambridge, United Kingdom, 2019).
- “Consent Decree of Permanent Injunction,” Ranbaxy Laboratories Ltd. & Ranbaxy, Inc. (Princeton, New Jersey, 2012).
- “Form 483 Observations,” Able Laboratories (Cranbury, New Jersey, 2005).
- MHRA GMP, Data Integrity Definitions and Guidance for Industry 2nd ed. (Medicines and Healthcare products Regulatory Agency, London, United Kingdom, 2015).
- MHRA, GXP Data Integrity Guidance and Definitions (Medicines and Healthcare products Regulatory Agency, London, United Kingdom, 2018).
- WHO, Technical Report Series No.996 Annex 5 Guidance on Good Data and Records Management Practices (World Health Organization, Geneva, Switzerland, 2016).
- FDA, Guidance for Industry Data Integrity and Compliance With Drug CGMP Questions and Answers (FDA, Silver Spring, Maryland, 2018).
- International Conference on Harmonization, PIC/S PI-041-3, Good Practices for Data Management and Integrity in Regulated GMP / GDP Environments Draft (ICH, Geneva, Switzerland, 2018).
- R.D. McDowall, Spectroscopy 31(4), 15–25 (2016).
- M.E.Newton and R.D.McDowall, LCGC North America 36(1), 46–51 (2018).
- R.D.McDowall, LCGC North America 37(1), 44–51 (2019).
- ISPE, Good Manufacturing Practice (GMP) Guidelines, chapter 1 (Pharmaceutical Quality System, European Commission, Brussels, Belgium, 2013).
- Current Good Manufacturing Practice for Finished Pharmaceutical Products, in 21 CFR 58 (Food and Drug Administration, Washington, D.C., 1978).
- OECD, Series on Principles of Good Laboratory Practice and Compliance Monitoring Number 1, OECD Principles on Good Laboratory Practice (Organization for Economic Co-operation and Development, Paris, France, 1998).
- Current Good Manufacturing Practices, in Good Guidance Practices, Level 2 Guidance - Records and Reports. (U.S. Government Printing Office, Washington, D.C., 2010).
- FDA, Cetero Research Untitled Letter (11-HFD-45-07-02) (FDA, Silver Spring, Maryland, 2011).
- US Food and Drug Administration, Letter to ANDA Sponsors Conducting Bioequivalence Studies at Cetero Research (FDA, Rockville, Maryland, 2011).



R.D. McDowall is the director of R.D. McDowall Limited and the editor of the “Questions of Quality” column for LCGC Europe, Spectroscopy’s sister magazine. Direct correspondence to: SpectroscopyEdit@MMHGroup.com











Synthesizing Synthetic Oligonucleotides: An Interview with the CEO of Oligo Factory
February 6th 2024LCGC and Spectroscopy Editor Patrick Lavery spoke with Oligo Factory CEO Chris Boggess about the company’s recently attained compliance with Good Manufacturing Practice (GMP) International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Expert Working Group (Q7) guidance and its distinction from Research Use Only (RUO) and International Organization for Standardization (ISO) 13485 designations.



