Electron-Capture Dissociation in a Radio-Frequency Linear Ion Trap
Special Issues
Here we describe a new compact device for electron-capture dissociation (ECD) analysis of large peptides and posttranslational modifications of proteins, which can be difficult to analyze via conventional dissociation techniques such as collision-induced dissociation (CID). The new compact device realizes ECD in a radio frequency (RF) linear ion trap equipped with a small permanent magnet, which is significantly different than the large and maintenance-intensive superconducting magnet required for conventional ECD in Fourier-transform ion cyclotron resonance mass spectrometers. In addition to its compactness and ease of operation, an additional merit of an RF linear ion trap ECD is that its reaction speed is fast, comparable to CID, enabling data acquisition on the liquid-chromatography (LC) time scale. We interfaced the linear-trap ECD device to a time-of-flight mass spectrometer to obtain ECD spectra of phosphorylated peptides injected into a liquid chromatograph, infused glycopeptides, and intact small..
Here we describe a new compact device for electron-capture dissociation (ECD) analysis of large peptides and posttranslational modifications of proteins, which can be difficult to analyze via conventional dissociation techniques such as collision-induced dissociation (CID). The new compact device realizes ECD in a radio frequency (RF) linear ion trap equipped with a small permanent magnet, which is significantly different than the large and maintenance-intensive superconducting magnet required for conventional ECD in Fourier-transform ion cyclotron resonance mass spectrometers. In addition to its compactness and ease of operation, an additional merit of an RF linear ion trap ECD is that its reaction speed is fast, comparable to CID, enabling data acquisition on the liquid-chromatography (LC) time scale. We interfaced the linear-trap ECD device to a time-of-flight mass spectrometer to obtain ECD spectra of phosphorylated peptides injected into a liquid chromatograph, infused glycopeptides, and intact small proteins. The results demonstrate that the RF linear trap ECD mass spectrometer has the potential to analyze posttranslational modifications and to perform de novo sequencing of peptides and small proteins with LC-compatible high-throughput performance.
Dissociation of peptide and protein ions is a key technique in proteomic studies using a mass spectrometer because dissociation products, or fragment ions, give information about the peptide sequence and posttranslational modifications (PTM) of the peptides and proteins. The widely used conventional dissociation technique is collision-induced dissociation (CID), which cleaves peptide bonds of the ions by collisionally exciting the internal energy of ions with neutral gas molecules. CID has a few disadvantages caused by its collisional process: collisions cannot deliver sufficient energy to all the peptide bonds for larger peptides, resulting in incomplete sequence information, and many types of PTMs are lost during the collisional process, complicating the determination of the modification location.
Electron-capture dissociation (ECD), a completely different dissociation method compared to CID, reduces positive charges via electron capture and induces the cleavage of multiply protonated peptides and proteins (Figure 1). The utility of this dissociation technique has become widely recognized as a potentially powerful tool for proteomics. In ECD, which was demonstrated initially in an FT-ion cyclotron resonance (FT-ICR) mass spectrometer (1), free electrons traveling in vacuum are captured by precursor ions. Low-energy electrons, whose kinetic energy is less than 1 eV, are indispensable for efficient electron capture. Another dissociation method similar to ECD, which uses charge reduction by electron transfer, is electron transfer dissociation (ETD). In ETD, first reported in 2004 (2), electrons are provided from negatively charged reagent molecular ions.


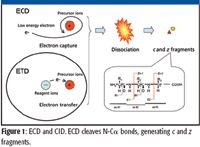
Figure 1
Both charge-reduction dissociation techniques, ECD and ETD, randomly cleave N-Cα bonds of the peptide backbone regardless of the type of amino acid residues on the N- and C-terminal sides of each bond; the one exception is the N-terminal side of proline. The cleavage is without apparent thermal excitation of the vibrational and rotational states. These two unique characteristics provide the following advantages for proteomics: evenly distributed cleavage sites over the entire peptide backbone bonds, even for large peptides and proteins, yielding confident matches and enabling the analysis of intact proteins, and efficient PTM analysis due to the preservation of the modifications on the peptides–proteins after cleavage, allowing for relatively easy identification of the modification site.
Although ECD has been recognized as a powerful tool for proteomics since its first demonstration in 1998, to date, its use has been limited to a static electromagnetic ion trap in an FT-ICR MS system. It is evident that proteomics can benefit further if the merits of ECD can be pursued in more common platforms such as ion traps and time-of-flight (TOF) systems. Furthermore, reported LC–ECD experiments using an FT-ICR MS system (3) show a time scale of tens of seconds for ECD spectral acquisition, suggesting that improved ECD speed is desirable for LC–MS applications. Potentially, RF ion traps can serve as a platform to realize a high-throughput, relatively inexpensive ECD device that is easy to operate and maintain. However, this has proven impossible to realize until recently because of the difficulty of introducing low-energy electrons into a strong RF field with an amplitude of 100–1000 V. In 2004, two groups accomplished this by focusing low energy electrons using a magnetic field together with an RF electric field; ECD was demonstrated in an RF linear ion trap by Baba and colleagues (4) and in a Paul-type quadrupole ion trap by Silivra and colleagues (5). Another approach using a digital RF ion trap without a magnetic field also was demonstrated by Ding and colleagues in 2005 (6).
ECD in a Radio-Frequency Ion Trap
The RF ECD device is composed of a linear rf-quadrupole ion trap with a cylindrical permanent magnet and an electron-beam source (4), as shown in Figure 2. Both precursor and product ions are trapped by the linear RF field in the radial direction and a dc electric field generated by a bias on the wall electrodes in the axial direction. For ECD to be efficient, the heating of the electron beam produced by the electron source must be minimized because it significantly decreases the electron capture efficiency. The magnetic field confines the low energy electron beam to travel along the central axis; along the axis there is no RF heating.


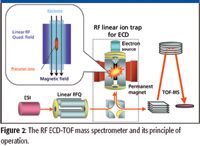
Figure 2
In the original demonstration of RF ECD (4), the ECD reaction speed was very slow due to inefficient ion and electron transmissions in the RF ECD device. This was improved drastically by a T-shaped arrangement, shown in Figure 2 (7). This arrangement, with increased ion and electron transmission efficiencies, resulted in more than four orders of magnitude improvement in the ECD reaction speed. The efficiency of ion transmission improved because the ions were injected and ejected from a different location than the electrons. This arrangement avoids field deformation resulting from electrode contamination via electron bombardment. The electron transmission improved because of the straight-entry configuration. As a result, the ECD reaction speed is now comparable to that of the conventional high-throughput dissociation method, CID. This, to the best of our knowledge, is more than two orders-of-magnitude faster than reported ECD experiments in an FT-ICR system (8).
ECD-TOF MS
The RF ECD device was installed in a modified linear ion-trap TOF mass spectrometer, the Hitachi NanoFrontier (Central Research Laboratory, Hitachi, Ltd., Japan). Multiply protonated precursor ions are produced via a nanoflow electrospray ionization source and isolated by the linear ion trap in the mass spectrometer. The isolated precursor ions are introduced into the RF ECD device for dissociation. The resulting ECD product ions are introduced into a TOF mass spectrometer for mass spectral acquisition.
Results
ECD Reaction Speed and Efficiency
Performance of the RF ECD was verified using doubly protonated substance P as a precursor ion. The electron capture rate is proportional to the square of the charge state of the precursor ions (1); therefore, the doubly charged ion provides the lowest reaction speed for substance P. Figure 3 shows a typical ECD spectrum for doubly charged substance P. The precursor ions were accumulated for 10 ms and isolated. Then, during the ECD procedure, a 0.5-mA electron beam was irradiated for 10 ms and the product ions were analyzed by TOF MS. This event was repeated for 0.5 s to accumulate the ECD spectrum shown in the figure (sample consumption of 1.4 fmol). The spectral peak series of c fragments and charge-reduced precursor ions indicated by [M+2H]+ are clearly observed.



Figure 3
When the duration time of the electron-beam irradiation for ECD reaction was varied from a few milliseconds to 40 ms, the intensity of remaining precursor ions decayed exponentially with a 1/e decay time of 5 ms; this indicates the electron-capture rate of the doubly charged ion. The total ECD product intensity increased for the first 7 ms and subsequently decreased exponentially with a 1/e decay time of 15 ms. This exponential decay of product ions after the vanishing of the precursor ions corresponds to the electron capture rate of the singly charged ECD product ions. Therefore, a fast electron capture of 10-ms time scale was accomplished; this is in the same range as CID. For multiply charged precursor ions, the dissociation speed would be faster than that for the doubly charged ion presented here.
The observed peak intensity of the ECD product ions over the initial precursor ion intensity was 50%. This is almost identical to the theoretical limit of the fragment observation efficiency given by a simple double exponential decay model whose decay rates were given by Z2. The RF ECD system presented here can be optimized for speed and efficiency comparable to that of CID.
LC Injection of Phosphorylated Peptides
Phosphorylation, which regulates cell cycles and cell-to-cell signal transduction, is one of the most important PTMs related to disease mechanisms. It is known that conventional CID of phosphorylated peptides induces neutral loss of phosphoric acids, complicating identification of the sequence and the location of phosphorylation (9), which becomes increasingly more difficult for multiply phosphorylated peptides.
Figure 4 shows RF ECD spectra of two phosphorylated peptides contained in a tryptic digest of bovine β casein digest. The tryptic digest was injected into a nano LC coupled to the RF ECD-TOF MS. The LC flow rate was 50 nL/min for an elution time of 70 min by a nanoflow pump (Hitachi High Technologies). The acquisition time for a single ECD spectrum was 4 s.



Figure 4
The peptide shown in the top of Figure 4 appeared at a retention time of 50 min, at which quadruply charged precursor ions were isolated and dissociated by ECD. All four phosphorylated sites were identified with no loss of phosphorylation — these sites typically are cleaved from the peptides when CID is applied. An additional phosphorylated peptide, shown in the bottom of Figure 4, appeared at a retention time of 37 min. The triply charged precursor ion of this peptide was isolated and dissociated. The singly phosphorylated peptide was sequenced easily without loss of the modification.
Infused Glycosylated Peptides
Glycosylation is another important PTM of proteins in biological, pharmaceutical, and medical applications. Figure 5 shows ECD spectra of N-linked and O-linked glycopeptides obtained by RF ECD-TOF MS. The top figure shows an ECD spectrum for an N-linked glycopeptide from chicken egg yolk. The glycan was preserved on the peptide after dissociation and the modification site was identified. The bottom figure shows an RF ECD spectrum of an O-linked glycopeptide with a sialic acid moiety (10). O-linked glycopeptides pose further challenges compared with N-linked glycopeptides. The sialic acid, which is known to be extremely labile and is removed easily from the glycan, was preserved on the glycopeptides after the ECD. Both the peptide sequence and the modification site were identified.


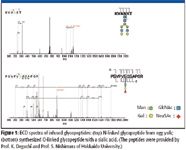
Figure 5
Analysis of an Intact Protein
Analysis of intact proteins is another important application of ECD. Figure 4 shows an RF ECD spectrum of intact ubiquitin (76 aa) that was obtained by irradiating electrons for 5 ms duration for ECD reaction. ECD fragments with charge states (Z) from 1 to 5 were identified, and the total number of identified sites was 65 out of a total of 75 cleavage sites. The largest identified fragment in the spectrum is c62, which was characterized by m = 1400 and Z = 5. This demonstrates that RF ECD-TOF MS has the potential to identify proteins with a mass of 14 kDa in a top-down, de novo manner.



Figure 6
Conclusion
We have developed a fast ECD device using a rf-quadrupole ion trap with a magnetic field. The reaction speed of the presented RF ECD system is comparable to that of the conventional dissociation technique, CID. LC-injected phosphorylated peptides, infused glycopeptides, and small proteins were analyzed via the RF ECD-TOF mass spectrometer. This demonstrates that the RF ECD-TOF system has the potential for high-throughput, high-performance analysis of PTMs and de novo sequencing ranging from peptides to small intact proteins.
Acknowledgment
The authors are grateful to Prof. Kisaburo Deguchi and Prof. Shinichiro Nishimura of Hokkaido University (Hokkaido, Japan) for their valuable discussions. The ECD project was partially supported by the New Energy and Industrial Technology Development Organization (NEDO), Japan.
NanoFrontier is a registered trademark of Hitachi High Technologies Corporation.
Takashi Baba, Hiroyuki Satake, Naomi Manri, Hideki Hasegawa, Atsumu Hirabayashi, and Yuichiro Hashimoto are with Central Research Laboratory, Hitachi, Ltd., Japan.
Izumi Waki is with Hitachi High Technologies, Hitachinaka, Ibaraki, Japan.
Chad Ostrander is with Hitachi High Technologies America, Dallas, Texas.
References
(1) R.A. Zubarev, N.L. Kelleher, and F.W. McLafferty, J. Am. Chem. Soc. 120, 3265 (1998).
(2) J.E. Syla, J.J. Coon, M.J. Schroeder, J. Shabanwitz, and D. Hunt, Proc. Natl. Acad. Sci. USA 101, 9528 (2004).
(3) A.J. Creese and H.J. Cooper, J. Am. Soc. Mass Spectrom. 18 891 (2007).
(4) T. Baba, Y. Hashimoto, H. Hasegawa, A. Hirabayashi, and I. Waki, Anal. Chem. 76, 4263 (2004).
(5) O.A. Silivra, F. Kjeldsen, I.A. Ivonin, and R.A. Zubarev, J. Am. Soc. Mass Spectrom. 16, 22 (2005).
(6) L. Ding and F.L. Brancia, Anal. Chem. 78, 1995 (2006).
(7) American Society of Mass Spectrometry Conference (2005 poster #TP12-238, 2006 oral # WOB11:15, and 2007 poster # MPF100, TPB023, TPP277).
(8) R.A. Zubarev, E.K. Fridriksson, D.M. Horn, N.L. Kelleher, N.A. Kruger, B.K. Carpenter, and F.W. McLafferty, Anal. Chem. 72, 563 (2000).
(9) A. Stensballe, O.N. Jensen, J.V. Olsen, K.F. Heselmann, and R.A. Zubarev, Rapid Commun. Mass Spectrom. 14, 1793 (2000).
(10) K. Deguchi, H. Ito, T. Baba, A. Hirabayashi, H. Nakagawa, M. Fumoto, H. Hinou, and S. Nishimura, Rapid Commun. Mass Spectrom. 21, 691 (2007).

















Trending on Spectroscopy: The Top Content of 2024
December 30th 2024In 2024, we launched multiple content series, covered major conferences, presented two awards, and continued our monthly Analytically Speaking episodes. Below, you'll find a selection of the most popular content from Spectroscopy over the past year.


Best of the Week: Hyperspectral Imaging, ICP-MS Analysis of Geological Samples, Product Roundup
October 18th 2024Top articles published this week include an article about hyperspectral imaging in human skin research, a peer-reviewed article about analyzing geological samples using atomic spectroscopy techniques, and an equipment roundup piece about the latest products in the industry.

