An Integrated Risk Assessment for Analytical Instruments and Computerized Laboratory Systems
A risk assessment helps determine the amount of qualification and validation work necessary to show that instruments and computerized laboratory systems are fit for their intended purpose. Here's how to do it.



A risk assessment is presented for determining the amount of qualification and validation work required to show that instruments and computerized laboratory systems are fit for their intended purpose.
Risk management is one of the new requirements for the pharmaceutical industry following the publication of the Food and Drug Administration's (FDA) "Good Manufacturing Practices (GMPs) for the 21st Century" (1) and the International Conference on Harmonization (ICH) Q9 on Quality Risk Management (2). How much qualification and validation work is required in connection with a regulated task is dependent on a justified and documented risk assessment. The United States Pharmacopeia (USP) General Chapter <1058> (3) on analytical instrument qualification (AIQ) has an implicit risk assessment in that it classifies instrumentation used in a regulated laboratory into one of three groups: A, B, or C. The chapter defines the criteria for each group, but leaves it to individuals to decide how to operate the classification in their own laboratories.
Software is pervasive throughout the instruments and systems in groups B and C, as acknowledged by <1058> (3). From a software perspective, Good Automated Manufacturing Practice (GAMP) 5 Good Practice Guide (GPG) for Validation of Laboratory Computerized Systems (4) is widely recognized within the industry and by regulators, but it is not consistent with some of the elements of USP <1058>. The USP general chapter is currently under revision and ideally the revised version will be fully compatibility with the GAMP 5 guidelines and good practice guides (4–6). In the meantime, however, users are left with a question: Do I follow USP <1058> or GAMP 5? We shall answer this question here.
Some Problems with USP <1058>
In November 2010, there was an American Association of Pharmaceutical Scientists (AAPS) meeting in New Orleans, Louisiana, where the status of <1058> was debated. That same month Bob published some of his thoughts about the advantages and disadvantages of that general chapter (7). The advantages consisted of the classification of instruments and systems, which was also its greatest disadvantage, as it was too simplistic. Simply saying that an instrument fit into group B ignored the possibility that there were in-built calculations that needed to be verified because of 21CFR 211.68(b) requirements (8) or that some instruments enable users to build their own programs. Furthermore, the approach to software for group C systems was naïve as it placed the responsibility for validation on the supplier rather than the user. Chapter <1058> also referenced the FDA guidance for industry entitled General Principles of Software Validation (9), which was written primarily for medical devices; the configuration and customization of software is not mentioned there.
GAMP Good Practice Guide for Laboratory Systems Updated
The publication of the first edition of the GAMP Good Practice Guide for Validation of Laboratory Computerized Systems (5) had some problems. However, in the recently published second edition (6), the good practice guide was aligned with GAMP 5 (4) and was updated to be risk-based (as reflected in the new title). Collaboration with us during the writing enabled both the good practice guide and the new draft of USP <1058> (11) to be more closely aligned and have a unified approach to qualification and validation of instruments and computerized laboratory systems. (The GAMP GPG uses the term laboratory computerized system in contrast to the more common term of computerized laboratory system; however the two terms are equivalent.) We have a paper soon to be published that maps the two approaches and shows that they are very similar despite some differences in terminology (12).
Progress Updating USP <1058>
The original basis of USP <1058> was the 2004 AAPS white paper "Analytical Instrument Qualification," which focused on a risk-based approach to AIQ by classifying apparatus, instruments, and systems depending on the laboratory's intended use. The definition of the intended use is the key part of the process, because the same item could be classified in any of the three groups depending on its use. Intended use is also an essential part of our risk assessment presented in this column.
However, the current weakness of the overall risk-based approach is the way in which software is assessed. Software is pervasive in group B instruments and group C systems. Chapter <1058> currently references the FDA guidance document, General Principles of Software Validation (9). This guidance was written primarily for medical device software, which is neither configured (modified to the business process by vendor supplied tools) nor customized (writing software macros or modules that are integrated with the application). Given that many analytical instruments and systems are configured or customized, this guidance does not fit well in a regulated GxP laboratory environment.
In January 2012, we published a stimulus to the revision process in the on-line version of Pharmacopeial Forum (13), in which we proposed an update for USP <1058>. In our proposal, instrument qualification was integrated with computerized system validation rather than being two separate activities. This would provide regulated laboratories with the opportunity to reduce the amount of work and avoid potential duplication. In this publication, we included a risk-assessment flow chart for determining the amount of work to perform to qualify analytical instruments and, where appropriate, validate the software functions and applications. From the comments received, we updated the flow chart. We present it here as a simplified method for classifying the apparatus, instruments, and systems in your laboratory.
Why an Integrated Risk Assessment Approach?
The basic risk assessment model in <1058> is the classification of any item used in a laboratory into group A, B, or C based on a definition of intended use. This is generally a sound approach, because apparatus (group A), instruments (B), or systems (C) are easily classified. However, there is a weakness in that the level of granularity currently offered by <1058> is insufficient to classify the variety and permutations of instruments (B) and systems (C) used in combination with software in the laboratory today.
Therefore the risk assessment presented in this column is to provide a means of
1. Unambiguously differentiating between apparatus (group A) and instruments (group B) based on functionality.
2. Linking software elements with the various types of instrument (group B) and systems (group C) as current instrumentation is more complex that the simplistic use of groups B and C in the current version of USP <1058>. This will identify subgroups within groups B and C.
Item 2 is a fundamental nonpedantic difference and is necessary for determining the proper extent of qualification or validation for a specific instrument or system. Effective risk management ensures that the appropriate amount of qualification and validation is performed relative to the stated intended use of an instrument or system. It does not leave a compliance gap for an inspector or auditor to find. Furthermore, verification of calculations is a necessary requirement of complying with US GMP regulations, specifically 21CFR 21.68(b) (8). This is omitted in the current version of <1058>.
Subdivision of Groups B and C
As mentioned earlier in this column, software is pervasive throughout the instruments in group B and systems in group C.
It is our contention that subgroups exist within groups B and C due solely to the software present and how it is used. The subgroups in groups B and C are shown in Table I.


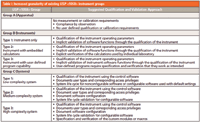
Table I: Increased granularity of existing USP instrument groups
We stress that this subclassification is important only when these features are used (that is, intended use). Otherwise, if the features are present but not used, then they are not relevant to the overall qualification or validation approach for the instrument or system. The issue then becomes one of system scope creep; because of that, the intended use of a given instrument needs to be reviewed regularly to ensure that it is still current. This is best undertaken during a periodic review.
Approach to Risk Assessment
We propose that a simple hierarchical risk assessment model be used to determine the extent of qualification or validation required to ensure "fitness for purpose" under the actual conditions of use within the laboratory. Our proposed model splits the USP <1058> groups B and C categories into three subcategories each, as is shown in Table I.
Integrated Risk Assessment Approach
The fundamental idea is to provide an unambiguous assignment of category that is documented in a qualification and validation statement that is completed at the end of the risk assessment. The process flow for this risk-assessment model is based on 15 simple, closed (yes or no) questions around six decision points shown in Figure 1.


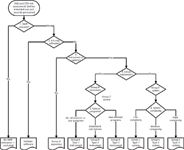
Figure 1: Schematic of an integrated analytical instrument qualificationâcomputerized system validation (AIQâCSV) risk assessment.
This process flow is simpler than one we originally published (11) and therefore it is easier to understand. The process consists of a preparation phase followed by six decision stages and is completed by a sign off by the owner with quality unit or quality assurance approval. If required, this risk assessment can be adapted easily to focus only on groups A, B, and C to exclusion of nonGMP relevant instruments and software; the choice is yours. However, we have chosen to present a comprehensive risk assessment to cover the majority of items used in a regulated laboratory.
Assessment Preparation: Describe Item and Define Intended Use
Preparing for a risk assessment is an administrative process and begins with describing the item using supplier, model, serial number, software name, and version or firmware version (as applicable). If required you can also add the owner and, if appropriate, the department and location of the item as well as an inventory or asset number if available.
The next, and most important, part of this preparation phase is to describe the intended use of the item. This is the key to the whole risk assessment. It is essential to be as accurate as possible and also to indicate if there are any records created by the use of the item (for example, paper printouts or electronic records). The intended-use statement should also indicate if the item will be connected to the network or will stand alone.
Some people may consider simplifying the intended use statement, but this is the most important part of this stage. Failure to define the intended use adequately means that the only person who is fooled is you. Ensure that the intended use is defined well. For example, if an instrument has 90% of its work involved in research and only 10% with GxP work, it is natural to focus on the research element. However, it is the 10% of the GxP work that is critical and determines the overall level of control required.
Step 1: Determine GMP Relevance
The first stage of the risk assessment is to determine if the item is carrying out GMP work. For the sake of completeness of the risk-assessment model we have included the possibility of laboratory instrumentation and systems being present that are not used for regulatory purposes. Based on the intended-use statement from the preparation stage, we ask the six closed questions (questions 1–6). These have been taken from the Society for Quality Assurance (SQA) Computer Validation Initiative Committee (CVIC) risk assessment questionnaire (14). This questionnaire posed 15 closed questions to determine if a computerized system carried out any GxP activities. We have taken only the relevant laboratory questions and used them in this <1058> risk assessment.
The questions asked are related to what the item is used for. Is the item used for
1. Testing of drug product or API for formal release?
2. Shipment of material, such as data loggers?
3. Nonclinical laboratory studies intended for submission in a regulatory dossier?
4. Clinical investigations including supply of clinical supplies or pharmacokinetics?
5. Generation of, submissions to, or withdrawal of a regulatory dossier?
6. Backup, storage, or transfer of electronic records supporting any of the above?
If all responses are "no" then qualification and validation are not necessary because the item has no GxP function and is documented as requiring no qualification or validation. This is highly unlikely to happen within a GMP-regulated quality control laboratory. However, if the laboratory is on the boundary between research and development there may be some interesting issues to manage. If the majority of work is carried out for research and a minority of the work for development (say a 90:10 split of activities) do we focus on the 90% or the 10%? Because there is a high probability that the 10% will end up in a regulatory dossier, we need to focus on the 10% to ensure the instrument and any software output are correct. Furthermore, if procedures developed using the instrument or system are to be transferred to other laboratories later in the lifecycle, we need to ensure the item is under control and the output is verified.
Step 2: Is the Item Standalone Software?
At step 2 only one closed question is asked (question 7) to determine if the item is standalone software:
7. Is the item only software that performs a GxP function or creates GxP records?
This question should be applied not only to recognized software applications such as statistical software or a laboratory information management system (LIMS), but also Excel (Microsoft) spreadsheets and Access (Microsoft) databases used within the laboratory. If the answer to question 7 is "yes," then the organization's validation procedures for computerized systems should be followed. Again, this question is included to ensure completeness of coverage of the risk assessment questionnaire for all laboratory instruments, systems, and software.
If the answer to question 7 is "no," then it is necessary to determine if the item is apparatus (group A) as opposed to instruments and systems (USP <1058> group B or group C).
Step 3: Is the Item an Apparatus (in Group A)?
The third step in the risk assessment is to differentiate between apparatus and instrumentation or systems. USP <1058> defines group A as standard apparatus with no measurement capability or user requirement for calibration (3). Therefore, we ask three closed questions (questions 8–10) to identify items of apparatus:
8. Is there any measurement capability of the item?
9. Is the item user calibrated after purchase?
10. Does the use of the item require more than observation?
If the answer to each question is no, then the item is classified as USP <1058> group A. On the other hand, if one or more answers are yes, then we move to step 4.
Step 4: Is the Item an Instrument (in Group B) or a System (in Group C)?
Group B includes standard equipment and instruments providing measured values as well as equipment controlling physical parameters (such as temperature, pressure, or flow) that need calibration. In contrast, group C systems include instruments and computerized analytical systems, where user requirements for functionality, operational, and performance limits are specific for the analytical application. These systems typically are controlled by a standalone computer with specific software for instrument control and data acquisition and analysis (3). To determine which items belong in these two groups, at step 4 two closed questions are asked (questions 11 and 12):
11. Does the item measure values or control physical parameters requiring calibration?
12. Is there a separate computer for control of an instrument and data acquisition?
If both answers are "yes" then the item is considered a system and falls under USP <1058> group C. However, if the answer to question 12 is "no" then the item is considered an instrument and falls under USP <1058> group B. The exact group B instrument subclassification (I, II, or III) is determined by asking three further closed questions regarding customization and configuration in step 5.
Step 5: Group B Instrument Subclassification
At step 5 the three remaining questions are asked (questions 13–15) to determine the subcategory of group B that the instrument belongs to (see Table I). The closed questions are as follows:
13. Are there any built-in calculations used by the instrument that cannot be changed or configured?
14. Can you configure the built-in calculations in the instrument?
15. Can you write a user-defined program with the instrument?
Group B, Type 1 instruments are indicated only if question 13 is answered "yes" because this is an instrument without any calculations or the ability for the user to define programs. Therefore, only qualification of the instrument's functions is required to demonstrate its intended purpose.
Group B, Type 2 instruments are designated if the answers to questions 13 and 14 are "yes" and to question 15 is "no." Therefore, in addition to qualification of the instrument the embedded calculations need to be verified in the way that they are used by the laboratory.
Group B, Type 3 instruments are classified if the answer to Q15 is "yes" and the remaining two answers are "no." This type of instrument requires qualification of the instrument plus specification and verification of any user defined programs. The latter process could be controlled by a standard operating procedure so that user-defined programs developed after the initial qualification can be validated on the operational system. Ideally, the user defined programs should be controlled to prevent change by unauthorized persons or without change control. It is possible that Type 3 instruments could also have embedded calculations, where the answer to Q14 was also "yes," which would result in these calculations being verified the same way as Type 2 instruments.
Let us return to the intended use statement in the preparation step discussed earlier. The instrument may be capable of performing embedded calculations or have the ability for users to define programs. However, if these functions are not intended to be used, then they do not need to be verified. Hence, you can see the importance of the intended use statement that was written in the preparation stage of the assessment.
Step 6: Group C System Subclassification
As noted in Table I, there are three types of group C systems that need to be differentiated. System complexity for systems in group C is determined by two factors. The first is the impact of the records generated by the system. Our risk assessment uses the scheme in the GAMP Good Practice Guide Compliant Part 11 Records and Signatures (see reference 15 for this classification). Second, the complexity of the system is determined by the nature of the software used to control the instrument and to acquire, process, store, and report results. Here, we use the GAMP 5 software classification (5).
Determination of Record Impact
The GAMP Part 11 guide (15) identifies three types of records generated in the pharmaceutical industry:
- High impact: Records typically have a direct impact on product quality, patient safety, or could be included in a regulatory submission — for example, batch release, stability studies, method validation records
- Medium impact: Records typically have an indirect impact on product quality or patient safety — for example, supporting records such as calibration, qualification, or validation records
- Low impact: Records typically have a negligible impact on product quality or patient safety and are used to support regulated activities but are not the key evidence of compliance — for example, plans for qualification or calibration activities (15)
Note that most of the records generated in a QC laboratory will typically be high impact. However, in an analytical development laboratory there may be more of a mixture of high- to low-impact records. In this case, the worst case scenario should be taken to avoid understating the risk offered by the system.
Determination of GAMP Software Category
The subclassification of USP <1058> group C systems is completed by determining the GAMP software category or categories that are used to control the instrument and acquire and process data from it (4). The three categories that we consider in this portion of the risk assessment are
- Category 3: Nonconfigurable software in which the software cannot be changed to alter the business process automated. There is limited configuration such as establishing and managing user types and the associated access privileges of each one, report headers, and location of data storage of records. However, this limited configuration does not change the business process automated by the software.
- Category 4: Configurable software that uses tools provided by the supplier to change the business process automated by the application. The tools can include entering a value into a field (such as reason for change within an audit trail), a button to turn a function on or off, or a supplier language. In the latter case, the "configuration" should be considered under custom software.
- Category 4 with category 5 custom modules or macros: In this case, we have the configurable software above, but with either custom modules programmed in a widely available language or macros for automating the operation of the application that are devised and written by the users using tools within the application. Category 5 is the highest risk software because it is typically unique to a laboratory or an organization. We will not consider custom applications in this risk assessment because the overwhelming majority of systems used in the regulated laboratory are commercially available applications that are either category 3 or 4.
Determination of Group C Complexity
When the impact of the records generated by the system and the software category or categories has been determined, it is compared with the grid in Figure 2 to determine the system complexity. This will also determine the amount of qualification and validation work required.


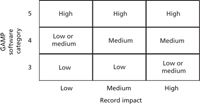
Figure 2: Subclassification of USP group C systems by software category and record impact.
As presented in Figure 2, there are three types of systems possible in group C:
- Group C, Type 1 system: A low complexity laboratory computerized system where the instrument is controlled by category 3 software that generates low-, medium-, or high-impact records or unconfigured category 4 software that generates low-impact records.
- Group C, Type 2 system: A medium complexity system consists of an instrument controlled by category 4 configurable software generating low-, medium-, or high-impact records. An alternative classification for a laboratory computerized system with category 3 software that generates high-impact records is that the system can be medium complexity.
- Group 3, Type 3 system: A high complexity system is one with an instrument controlled by category 4 software that has category 5 software modules or macros. It can generate low-, medium-, or high-impact records. It is the incorporation and use of the custom or category 5 software that makes the system high complexity, because this software is unique and needs more control.
The approach to the control of these systems is through a combination of qualification of the analytical instrument with the validation of the application software (that is, an integrated approach). The controlling document for this work is either a standard operating procedure (SOP) or a validation plan. Because of the wide variation of systems, the first option would be a validation plan for high and most medium complexity systems. For some medium and most low complexity systems, using an SOP for validation or qualification of simpler systems could be an easier and more efficient approach (16). Figure 2 can also be used in a different way, to identify the risk elements in the different software components of a system. Components with custom software require more control than those that are configured compared with those that are not configured.
The system life cycle to be followed and the documented evidence required for qualification of instruments and validation of the software should be defined in a validation plan or SOP. The details of this are outside the scope of this column, and readers should refer to existing approaches (4,6).
Summary
In this column we have presented a comprehensive risk assessment process for classifying apparatus, instruments, and computerized laboratory systems used in regulated laboratories. The intention is to demonstrate clearly that an integrated approach to analytical instrument qualification and, where appropriate, computerized system validation, is more efficient than separating the two tasks. The risk assessment also ensures that laboratories qualify or validate all intended use functions in an item and aims to minimize regulatory exposure from omitting work.
References
(1) US Food and Drug Administration, Guidance for Industry: Good Manufacturing Practices for the 21st Century (FDA, Rockville, Maryland, 2002).
(2) International Conference on Harmonization, ICH Q9, Quality Risk Management (ICH, Geneva, Switzerland, 2008).
(3) General Chapter <1058> "Analytical Instrument Qualification" in United States Pharmacopeia 36 –National Formulary 31 (United States Pharmacopeial Convention, Rockville, Maryland, 2012).
(4) ISPE, Good Automated Manufacturing Practice (GAMP) Guide, version 5 (International Society of Pharmaceutical Engineering, Tampa, Florida, 2008).
(5) ISPE, GAMP Good Practice Guide Validation of Laboratory Computerized Systems, 1st Edition (International Society of Pharmaceutical Engineering, Tampa, Florida, 2005).
(6) ISPE, GAMP Good Practice Guide: A Risk Based Validation of Laboratory Computerized Systems, 2nd Edition (International Society of Pharmaceutical Engineering, Tampa, Florida, 2012).
(7) R.D. McDowall, Spectroscopy 25 (11), 24–29 (2010).
(8) Current Good Manufacturing Practice Regulations for Finished Pharmaceutical Goods, in 21 CFR 211.68(b) (U.S. Government Printing Office, Washington, DC, 2008).
(9) US Food and Drug Administration, Guidance for Industry,General Principles of Software Validation (FDA, Rockville, Maryland, 2002).
(10) R.D. McDowall, Spectroscopy 26 (12), 14–17 (2012).
(11) Proposed revision to United States Pharmacopeia <1058> Analytical Instrument Qualification in United States Pharmacopeia (United States Pharmacopeial Convention, Rockville, Maryland, 2013).
(12) L. Schuessler, M.E. Newton, P. Smith, C. Burgess, and R.D. McDowall, Pharm. Tech., in press.
(13) C. Burgess and R.D. McDowall, Pharmacopeial Forum 38 (1), 2012.
(14) Computer Validation Initiative Committee of the Society of Quality Assurance, Computer Risk Assessment (Society of Quality Assurance, Charlottesville, Virginia, circa 1997).
(15) ISPE, GAMP Good Practice Guide, A Risk Based Approach to Compliant Electronic Records and Signatures, (International Society of Pharmaceutical Engineering, Tampa, Florida, 2005).
(16) R.D. McDowall, Quality Assurance Journal 12, 64–78 (2009).
R.D. McDowall is the Principal of McDowall Consulting and the director of R.D. McDowall Limited, and the editor of the "Questions of Quality" column for LCGC Europe, Spectroscopy's sister magazine. Direct correspondence to: spectroscopyedit@advanstar.com



R.D. McDowall
Chris Burgess is the Managing Director of Burgess Analytical Consultancy Limited, Barnard Castle, County Durham, UK. He has over 40 years experience as an analytical chemist including 20 years working in the pharmaceutical industry and 20 years as a consultant. Chris has been appointed to the United States Pharmacopoeia's Council of Experts 2010 to 2015 and is a member of the USP Expert Panel on Validation and Verification of Analytical Procedures.



Chris Burgess













