Life Science Applications of Electrochemistry Coupled to Liquid Chromatography–Mass Spectrometry
Redox reactions are integral parts of many cellular processes. Thus, they are extensively studied in vitro and in vivo. Electrochemistry (EC) represents a pure instrumental approach to characterize direct and indirect effects of redox reactions on bioorganic molecules.
Redox reactions are integral parts of many cellular processes. Thus, they are extensively studied in vitro and in vivo. Electrochemistry (EC) represents a pure instrumental approach to characterize direct and indirect effects of redox reactions on bioorganic molecules. EC may give rise to the formation of complex mixtures of intermediates, products, and by-products. Accordingly, efficient analytical techniques such as liquid chromatography–mass spectrometry (LC–MS) need to be applied for comprehensive characterization of the reaction mixtures obtained. Small molecules, peptides, proteins, and nucleic acids are common targets of EC–LC–MS studies, and this review is intended to give an overview on these important life science applications.
Redox reactions are essential for life. There are two important groups of biologically important redox reactions: enzymatic and nonenzymatic reactions. Enzymatic redox reactions often involve complex mechanisms of several enzymes. The electrons are transported by flavin- or heme-containing coenzymes from one reaction to another. These reactions represent an integral part of many metabolic pathways. Biological energy, for instance, is stored and released by means of redox reactions. Cellular respiration is the oxidation of glucose to carbon dioxide and the reduction of oxygen to water; photosynthesis involves the reduction of carbon dioxide into sugars and the oxidation of water into molecular oxygen. Furthermore, oxidoreductases such as the members of the cytochrome P450 superfamily generate structural complexity during natural product biosynthesis, and they play a central role in the biotransformation of drugs and toxins ("phase I metabolisms"). Biotransformation reactions involving xenobiotics are usually indented to make them more polar and, thus, enable a more rapid excretion. Biotranformation can lead to a loss or gain of activity. Furthermore, in some cases reactive intermediates are produced that can bind to proteins, lipids, and nucleic acids giving rise to cellular damage.
Biologically important redox reactions can also involve nonenzymatic processes. Reactive oxygen species (ROS) are natural by-products of the normal metabolism of oxygen. However, during times of environmental stress (for example, UV or heat exposure) ROS levels can increase dramatically. The availability of ROS can be important for an organism, as they are used by the immune system as a way to attack and kill pathogens. Furthermore, some ROS function as physiological regulators of intracellular signalling pathways. In the majority of cases, however, ROS production is considered harmful. ROS can react with proteins, lipids, and nucleic acids, which usually gives rise to cell damage called oxidative stress. In humans oxidative stress is thought to be involved in the pathogenesis of different kinds of disease, including neurodegenerative disease, cardiovascular disease, and cancer.
Because of their importance, biological redox reactions are extensively studied in in vitro and in vivo models. Electrochemistry (EC) was found to be a competent approach to complement existing assays in characterizing direct and indirect effects of redox reactions on bioorganic molecules. Electrochemistry was particularly useful in synthesizing oxidation products as well as reactive intermediates, which can be trapped by different kinds of electrophiles and nucleophiles. Moreover, electrochemistry is a purely instrumental approach. Thus, experimental conditions including the electrochemical potential, the electrode material, pH, as well as the kind and concentration of reactants, can precisely be controlled. Furthermore, electrochemistry is considered to be fast and automatable, which provides cost-savings.
Redox reactions may give rise to the formation of complex mixtures of intermediates, products, and by-products. Different kinds of analytical techniques can be used for comprehensive analysis. Particularly, mass spectrometry (MS) has found widespread application for that purpose. Mass spectrometric techniques enable qualitative (that is, identification and structure elucidation) and quantitative analysis. Among the different ionization techniques available, electrospray ionization (ESI) is the most commonly applied technique. ESI allows the MS characterization of a large variety of compounds ranging from small molecules to large biopolymers. The importance of EC–ESI-MS techniques is emphasized by the large number of reviews that have been published in recent years (1–13). Particularly in life sciences, EC–ESI-MS has found widespread application. The present review is intended to give a short overview on recent advances in that important field of research (Figure 1).


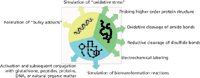
Figure 1: Summarizing sketch of the different kinds of applications that are realizable with ECâ(LC)ESI-MS in metabolomics, proteomics, and nucleic acids research.
Experimental Setup
Different experimental setups have been developed that enable EC–ESI-MS experiments. In the simplest setup, the inherent electrochemistry of ESI is used (12). From the electrochemical point of view (14–16), the ESI source represents a controlled-current cell consisting of two electrodes. One electrode is the capillary emitter; the mass spectrometer acts as the counter electrode. The two electrodes are connected on the one hand by the power supply and on the other hand by a series of resistors consisting of the electrochemical contact to the solution, the solution resistance, the resistance at the solution–air interface and in the gas phase, and the charge neutralization at the counter electrode. During operation, charges are transported from the emitter via the gas phase to the mass spectrometer. The loss of charges needs to be balanced in solution, and this is accomplished by electrochemical processes at the emitter electrode. These redox reactions may involve analytes (17–19), the solvent (20,21), or the electrode material (14,22). Practitioners have learned to control the electrochemical part of ESI in a way that the impact of electrochemistry on the mass spectra observed can be very much tuned. In the majority of cases, experimental conditions are chosen that prevent the mass spectrometric detection of ESI-inherent electrochemical reactions. However, in some situations electrochemistry can be used as an analytical advantage. A clear limitation of this setup is its inability to precisely control the electrochemical potential at the emitter electrode. Thus, experimental setups allowing separation of the electrochemical processes studied from the ESI-inherent electrochemistry are more commonly used.
Discrete electrochemical cells can be on-line hyphenated to ESI-MS (Figure 2a). The electrochemical cells used are typically controlled-potential cells consisting of three electrodes (that is, working electrode, reference electrode, and auxiliary electrode). The cells contain either porous flow-through or planar flow-by working electrodes. Cells with porous electrodes are considered to provide good conversion rates even at high flow rates because of the large surface area provided. Typically, glassy carbon is used as electrode material. A clear disadvantage of glassy carbon is the occurrence of analyte adsorption on the electrode surface. Thus, the cells are usually operated with solvents containing high organic contents. Other problems of this cell type are limited robustness and reproducibility. Life history or age of the electrochemical cell can sometimes have an impact on the oxidation reactions observed (23). For the planar thin-layer cells, adsorption on the working electrode is usually a less common problem (24). Another advantage of this cell type is the possibility to use different kinds of electrode materials. Besides glassy carbon, platinum and boron-doped diamond electrodes are the most promising alternatives. For obtaining high conversion rates, thin-layer cells are usually operated at very low flow rates (<10 µL/min) (25).


Figure 2: Commonly used setups for combining electrochemistry with LC and ESI-MS: (a) ECâMS, (b) LCâECâMS, (c) ECâLCâMS.
Very recently, electrochemical cells integrated in microfluidic devices were presented (26–28). Furthermore, configurations with electrochemical cells integrated into the ESI emitter are also available (29–31).
The coupling of a discrete electrochemical cell with the ESI-MS system is complicated by the need to decouple the cell voltage from the ESI high voltage (32). Decoupling is necessary for proper control of the electrochemical potential at the working electrode, and is usually accomplished either by using sufficiently long transfer lines (~30 cm) between the electrochemical cell and the ESI source (15,29), the insertion of a ground point between the electrochemical cell and the ESI source (29), or by allowing the whole system to float on the potential induced by the ESI high voltage (29).
For proper setup of an EC–ESI-MS system, sites for electrochemical reactions with limited user control should be kept to a minimum. Unwanted secondary reactions can take place at ground points, the ESI source, and auxiliary and reference electrodes (32–34). The mass transport to these sites is usually limited by using high solution flow rates, minimizing their accessible surface area, or avoiding any direct contact with the analyte in the flow stream (for example, separate electrode compartments).
The analytical power of EC–ESI-MS can be increased by integrating liquid chromatography (LC) as an additional dimension of separation. Chromatographic separation is particularly useful to reduce the complexity of the sample submitted either to the electrochemistry cell or to ESI-MS. Because of its compatibility with electrochemistry and ESI, reversed-phase chromatography is the preferred chromatographic mode of operation. The electrochemical cell can be integrated at different positions into the LC–MS system.
Postcolumn electrochemistry (Figure 2b) can be used to study the redox chemistry of selected compounds within complex mixtures (35,36). Pre-column electrochemistry (Figure 2c) enables the comprehensive characterization of redox reaction products via LC fractionation and subsequent MS detection (37,38). As the commonly applied chromatographic systems are operated at flow rates of several hundred microliters per minute, electrochemical cells with porous flow-through working electrodes with very high conversion efficiencies are exclusively used for both types of experimental setups. To enable the use of thin-layer cells, the electrochemistry cell can be integrated into the injection device (25,39). In such a setup, the sample solution is delivered through the electrochemical cell into the injection loop and subsequently transferred into the LC flow path.
Despite considerable success of the currently available electrochemistry instrumentation in converting bioorganic molecules, there is still a need for improved experimental conditions. A lot of research is going on to increase the capabilities, yield, and reproducibility of electrochemical reactions. The yield can be improved by optimization of reaction parameters including solvent composition, pH, and electrode material (25). Another strategy to increase the rate of conversion is based on the application of square-wave potential pulses (40,41). The superior performance with pulsing was attributed to an increased desorption of reaction products and continuous renewal of the electrode surface. The potential pulses were also found to give rise to the formation of products that were not seen in direct electrochemistry alone. Other approaches developed to expand the reactivity of electrochemistry include enzyme-modified systems (42) and hydroxyl radical-producing systems (43–46).
Metabolomics Applications
Based on the number of published articles, the most important field of application of EC–ESI-MS is studying the redox chemistry of small bioorganic molecules. In this context, EC–ESI-MS techniques are particularly useful to mimic biotransformation reactions.
Bruins and colleagues (47,48) were the first who systematically studied the redox reactivity of pharmaceutical compounds with EC–ESI-MS. This work laid the foundation for the use of EC–ESI-MS techniques to mimic phase I oxidative reactions in drug metabolism studies. Because valuable information concerning the sensitivity of a substrate toward oxidation can be obtained from EC–ESI-MS experiments, the technique is regarded as an efficient tool in the drug development process that complements existing in vitro and in vivo screening strategies. Electrochemistry was found to be particularly useful in cases where P450 enzyme-catalyzed reactions are supposed to proceed via a mechanism initiated by a one-electron oxidation. Typically, direct electrochemistry oxidation successfully mimics benzylic hydroxylation, hydroxylation of aromatic rings containing electron-donating groups, N-dealkylation, S-oxidation, dehydrogenation, and, less efficiently, N-oxidation and O-dealkylation. The range of oxidizable moieties was extended by the application of hydroxyl radical-producing systems (43–46). For instance, with electrochemically assisted Fenton chemistry aliphatic hydroxylation, aromatic hydroxylation, N-oxidation, and O-dealkylation were also efficiently mimicked. Furthermore, combining electrochemistry with enzyme-modified systems (42) holds the promise to enable simulation of the full range of biotransformation reactions occurring in vivo.
The applicability of EC–ESI-MS in drug metabolism studies has been demonstrated for a number of important pharmaceutical compounds, including amodiaquine (42,49–51), diclofenac (52,53), lidocaine (40,45,46), acetaminophen (49), haloperidol (54), troglitazone (55–57), clozapine (49), trimethoprim (49), flunitrazepam (58), clonazepam (58), chlorpromazine (58), zotepine (59,60), acebutolol (61,62), alprenolol (62), albendazole (63), tetrazepam (39), verapamil (64), and galantamin (65).
Phase I metabolism of drug compounds often gives rise to the formation of reactive species that are generally short-lived and unstable. These species can react with lipids, proteins, and nucleic acids giving rise to substantial damage. Within living organisms reactive intermediates are usually detoxified by binding to glutathione (phase II reaction). Reactive species are also produced in electrochemistry experiments. For the detection of these compounds and to mimic phase II metabolism, trapping agents are commonly applied. Appropriate nucleophiles include glutathione (51), glutathione derivatives (66), cysteine (67), and proteins (68). These compounds can be added to the sample solution after or before electrochemistry. The study of the skin-sensitizing potential of chemicals is a very interesting application of the ability of electrochemistry to produce electrophiles that subsequently react with proteins (69–71). Allergic reactions are most often caused by haptenation of proteins. A presupposition for this reaction is the ability of chemical allergens (small organic molecules and their metabolites) to form covalent bonds to nucleophilic sites in proteins (for example, cysteine and lysine). These activation and conjugation reactions can be studied in a well defined environment of selected nucleophiles with EC–ESI-MS, and this has been demonstrated for p-phenylendiamine, eugenol, and isoeugenol.
The study of biotransformation reactions involving pharmaceutical compounds is also gaining importance in environmental research. Xenobiotics are emitted from living organisms after biotransformation. Furthermore, biotransformation reactions may occur in the environment after exposure to terrestrial and aquatic microbial systems. Accordingly, comprehensive risk assessment of any chemical requires a detailed understanding of its different manifestations. Here, EC–ESI-MS can be used to produce putative metabolites as well as their conjugates with natural organic matter (72,73).
In comparison to drug metabolism studies, the application of EC–ESI-MS to study transformation reactions of endogenous compounds is less common. So far, there are only a few publications demonstrating the potential of this approach (37,38,74–77). Nevertheless, we believe that in the near future EC–ESI-MS will gain importance in this field of research. Electrochemistry reactions could become particularly useful to produce sufficient quantities of likely metabolites using known metabolites as reactants to enable their structural elucidation and subsequent confirmation in authentic samples by making use of spectral data stored in libraries or similar systems (78,79).
Proteomics Applications
In proteomics, there is a continuously growing interest in EC–ESI-MS. The technique can be used to study the oxidation of peptides and proteins as well as to selectively tag, label, or cleave these species.
Direct oxidation of peptides and proteins is widely studied, because of the role of oxidative damage in disease. The amino acids that most easily undergo oxidation are tyrosine, tryptophan, cysteine, methionine, and histidine (80). Tyrosine and tryptophan are hydroxylated. Cysteine and methionine are converted to their sulfonic acid and sulfone derivatives, respectively. Additionally, cysteines can form disulfide bonds, which may be oxidatively cleaved.
The direct oxidation of proteins can be used for analytical advantage to probe their higher order structure (81) by oxidizing solvent-exposed amino acid residues. Oxidization can be accomplished at physiologically relevant conditions and on-line monitored by MS. A clear drawback of the technique is that, even when only one protein is involved, complications such as low abundances across a range of different oxidized peptides, a wide variety of possible mass shifts, and fragmentation of coeluted oxidized peptide isoforms pose challenges to data interpretation, which currently renders electrochemistry more laborious and time-consuming than chemical approaches.
In 2003, Bruins and colleagues (80)realized that direct oxidation of proteins and peptides can induce cleavage of the amide bond at the C-terminal side of tyrosine and tryptophan residues. These cleavage reactions were observed in most analyzed peptides and proteins, which clearly suggested that electrochemistry could represent an instrumental alternative to chemical and enzymatic cleavage strategies. The main advantage of electrochemistry is the direct hyphenation to MS without the need to remove reagents, enzymes, or buffers. Thus, EC–ESI-MS enables direct cleavage and sequencing of proteins. The main problems encountered are related to yield, reproducibility, and adsorption phenomena. The cleavage rate can vary between different species and a considerable amount of noncleavage oxidation products are often observed (23,82). Nevertheless, the yield of cleavage products can be improved by proper control of the oxidation potential and the application of strongly acidic conditions (pH 2–3) (82). Furthermore, adsorption phenomena and electrode fouling can be reduced by changing from glassy carbon to boron-doped diamond electrodes and by applying regeneration procedures (83).
Direct reduction of peptides mainly targets disulfide bond bridges. Disulfide bonds are one of the most common post-translational modifications and provide covalent cross-linkages in native proteins for maintaining the three-dimensional structures of proteins and their biological activities. However, the presence of disulfide linkages increases the complexity for the protein structure elucidation by MS. Accordingly, they need to be reduced, and this is usually accomplished by chemical reduction. Recently, electrochemical reduction has been shown to represent a fast and efficient alternative for disulfide bond cleavage with immediate application in bottom-up and top-down proteomics (58,84–86). A clear advantage of electrochemistry is the possibility to analyze the reaction products directly by MS. Electrochemistry is a reagent-free cleavage approach. There is no need for any kind of sample preparation between cleavage and MS analysis. High conversion yields can be obtained with proper selection of experimental conditions (that is, Ti-based electrode, 1% formic acid in the solvent). In peptide mapping, electrochemistry enables the identification of disulfide-bridged peptides within enzymatic digest mixtures by inducing changes in ion abundance. Furthermore, in combination with tandem MS analysis, disulfide linkage pattern and sequence information for the examined peptides can be determined. At the protein level, electrochemical reduction of disulfide bonds enables tandem MS sequencing with high sequence coverage. Moreover, based on charge state distribution changes after electrolytic reduction, the role of disulfide bonds in maintaining protein folding can be evaluated.
Electrochemistry can also indirectly be used for peptide and protein characterization. Mass tags or labels for specific amino acid residues can be generated electrochemically (7,87–90). Usually, the inherent electrochemistry of ESI is applied for activation. Metal ions are used for noncovalent labeling, copper ions for targeting cysteine residues, and zinc ions for targeting histidine residues and phosphorylation sites. Covalent labeling usually involves activated hydroquinone species, which selectively react with cysteine residues. Electrochemically assisted labeling was found to be useful to determine the number of cysteine residues, which improves protein identification by database searching. Furthermore, based on the labeling extent of different residues within peptide and proteins, the accessibility, reactivity, or affinity of these sites can be tested. The usefulness of this approach has, for instance, been demonstrated by identifying the preferred binding sites of copper ions to β-amyloid 16, which is a protein that is involved in the development of Alzheimer's disease (91).
Applications in Nucleic Acids Research
DNA damage has emerged as a major culprit in cancer and many diseases related to aging. Oxidation reactions are typically involved in processes leading to DNA damage. Oxidative stress can lead to the production of ROS, which react with nucleic acids. Furthermore, exogenous chemical agents, such as toxins, pharmaceuticals, or pollutants, are often activated by oxidation to react with the genetic material. Produced lesions include base and sugar damages, strand breaks, crosslinks with proteins as well as the formation of bulky adducts. The cellular response to damage involves several processes such as DNA repair, cell cycle arrest, and apoptosis, while irreversible mutations contribute to oncogenesis.
To enable the development of strategies to protect the genetic material from oxidative stress, mechanistic aspects of nucleic acids oxidation have been extensively studied. The electroactivity of nucleic acids was discovered by Palecek in the 1960s (92). In the following years, a number of oxidation products of nucleobases, nucleosides, and nucleotides were identified using electrochemistry combined off-line with analytical techniques, such as ultraviolet–visible (UV–vis) spectroscopy, gas chromatography (GC)–MS after derivatization, or LC–MS (93–95). In this context, EC–ESI-MS holds the promise to facilitate and accelerate the identification process by eliminating laborious and time-consuming isolation and derivatization steps (25,76,96,97).
Particularly, EC–ESI-MS was applied to study the oxidation of guanine-containing species. Guanine exhibits the lowest oxidation potential among nucleic acids components and, therefore, is the preferential target of oxidation within nucleic acids. The primary products of guanine oxidation were identified as 8-hydroxyguanine and cross-linked guanines. The product 8-hydroxyguanine represents the most important biomarker to indicate oxidative damage of genetic material (98), and the formation of this compound clearly demonstrates that electrochemistry is able to mimic in vivo oxidation of nucleic acids induced by oxidative stress.
Figure 3 shows a comparison of the tandem mass spectra of dGpdG and electrochemically oxidized dGpdG. Because of electrochemical guanine oxidation, fragment ions appeared in the mass spectrum of the oxidized species that were not seen in the spectrum of dGpdG. Of particular importance for structure elucidation were w1 (G)- and w1 (oxoG)-. These ions prove that electrochemistry gave rise to the formation of a mixture of oxidation products having 8-hydroxyguanine either on the first or second position.


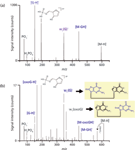
Figure 3: Comparison of the fragment ion mass spectra obtained from (a) dGpdG and (b) a mixture of its monooxidized forms. Electrochemical oxidation was performed on a boron-doped diamond electrode part of a thin-layer cell by applying a potential of 1.5 V. The electrochemical cell was on-line hyphenated to a quadrupole-quadrupole-time-of-flight instrument, where a collision voltage of -40 V was applied to initiate fragmentation. A more detailed description of the experimental conditions can be found in reference 97.
The formation of bulky adducts is an alternative mode of nucleic acids alteration (99). Here, oxidation reactions are involved in the activation processes; the produced electrophiles react with nucleophilic sites within the genetic material. EC-MS represented a useful tool to study the formation of adducts between nucleic acids and environmental pollutants (100), amino acids (101), and pharmaceutical compounds (97,102).
Conclusions
EC–ESI-MS represents a versatile tool for studying redox processes involving different kinds of bioorganic molecules. EC–ESI-MS is a purely instrumental approach. Electrochemistry enables the fast and automated generation of redox reaction products; ESI-MS, often in combination with LC separation, allows for the comprehensive characterization of the reaction mixture obtained. EC–ESI-MS represents a very useful complement to in vitro and in vivo techniques in metabolomics, proteomics, and nucleic acids research. Despite the considerable success of EC–ESI-MS in life sciences, further research efforts are necessary to increase its performance in terms of reproducibility, yield, and robustness.
Acknowledgments
This work was funded by the Austrian Science Fund (FWF): P 22526-B11. Furthermore, the authors want to thank Jean-Pierre Chervet (Antec, Zoeterwoude, The Netherlands) for his helpful discussions.
References
(1) G. Diehl and U. Karst, Anal. Bioanal. Chem. 373, 390–398 (2002).
(2) U. Karst, Angew. Chem. Int. Ed. Engl. 43, 2476–2478 (2004).
(3) W. Lohmann and U. Karst, Anal. Bioanal. Chem. 391, 79–96 (2008).
(4) A. Baumann and U. Karst, Expert Opin. Drug Metab. Toxicol. 6, 715–731 (2010).
(5) H. Faber, S. Jahn, J. Kunnemeyer, H. Simon, D. Melles, M. Vogel, and U. Karst, Angew. Chem. Int. Ed. Engl. 50, A52–A58 (2011).
(6) S. Jahn and U. Karst, J. Chromatogr. A 1259, 16–49 (2012).
(7) C. Roussel, L. Dayon, N. Lion, T.C. Rohner, J. Josserand, J.S. Rossier, H. Jensen, and H.H. Girault, J. Am. Soc. Mass Spectrom. 15, 1767–1779 (2004).
(8) H.P. Permentier, A.P. Bruins, and R. Bischoff, Mini Rev. Med. Chem. 8, 46–56 (2008).
(9) J. Roeser, R. Bischoff, A.P. Bruins, and H.P. Permentier, Anal. Bioanal. Chem. 397, 3441–3455 (2010).
(10) E. Nouri-Nigjeh, R. Bischoff, A.P. Bruins, and H.P. Permentier, Curr. Drug Metab. 12, 359–371 (2011).
(11) J.F. Mora, G.J. Van Berkel, C.G. Enke, R.B. Cole, M. Martinez-Sanchez, and J.B. Fenn, J. Mass Spectrom. 35, 939–952 (2000).
(12) G.J. Van Berkel and V. Kertesz, Anal. Chem. 79, 5510–5520 (2007).
(13) J. Gun, S. Bharathi, V. Gutkin, D. Rizkov, A. Voloshenko, R. Shelkov, S. Sladkevich, N. Kyi, M. Rona, Y. Wolanov, D. Rizkov, M. Koch, S. Mizrahi, P.V. Pridkhochenko, A. Modestov, and O. Lev, Isr. J. Chem. 50, 360–373 (2010).
(14) A.T. Blades, M.G. Ikonomou, and P. Kebarle, Anal. Chem. 63, 2109–2114 (1991).
(15) G.J. Van Berkel and F. Zhou, Anal. Chem. 67, 2916–2923 (1995).
(16) G.S. Jackson and C.G. Enke, Anal. Chem. 71, 3777–3784 (1999).
(17) G.J. Van Berkel, S.A. McLuckey, and G.L. Glish, Anal. Chem. 63, 1098–1109 (1991).
(18) X. Xu, S.P. Nolan, and R.B. Cole, Anal. Chem. 66, 119–125 (1994).
(19) R. Vessecchi, A.E.M. Crotti, T. Guaratini, P. Colepicolo, S.E. Galembeck, and N.P. Lopes, Mini-Rev. Organ. Chem. 4, 75–87 (2007).
(20) G.J. Van Berkel, F. Zhou, and J.T. Aronson, Int. J. Mass Spectrom. 162, 55–67 (1997).
(21) S. Plattner, R. Erb, J.P. Chervet, and H. Oberacher, Anal. Bioanal. Chem. 404, 1571–1579 (2012).
(22) G.J. Van Berkel, K.G. Asano, and P.D. Schnier, J. Am. Soc. Mass Spectrom. 12, 853–862 (2001).
(23) H.P. Permentier and A.P. Bruins, J. Am. Soc. Mass Spectrom. 15, 1707–1716 (2004).
(24) A. Baumann, W. Lohmann, T. Rose, K.C. Ahn, B.D. Hammock, U. Karst, and N.H. Schebb, Drug Metab. Dispos. 38, 2130–2138 (2010).
(25) R. Erb, S. Plattner, F. Pitterl, H.J. Brouwer, and H. Oberacher, Electrophoresis 33, 614–621 (2012).
(26) V. Mengeaud, O. Bagel, R. Ferrigno, H.H. Girault, and A. Haider, Lab Chip 2, 39–44 (2002).
(27) G. Liljegren, A. Dahlin, C. Zettersten, J. Bergquist, and L. Nyholm, Lab Chip 5, 1008–1016 (2005).
(28) M. Odijk, W. Olthuis, A. van den Berg, L. Qiao, and H. Girault, Anal. Chem. 84, 9176–9183 (2012).
(29) F. Zhou and G.J. Van Berkel, Anal. Chem. 67, 3643–3649 (1995).
(30) X. Xu, W. Lu, and R.B. Cole, Anal. Chem. 68, 4244–4253 (1996).
(31) G.J. Van Berkel, K.G. Asano, and M.C. Granger, Anal. Chem. 76, 1493–1499 (2004).
(32) C. Zettersten, R. Lomoth, L. Hammarstrom, P.J.R. Sjoberg, and L. Nyholm, J. Electro. Anal. Chem. 90, 90–99 (2006).
(33) H. Deng and G.J. Van Berkel, Electroanalysis 11, 857–865 (1999).
(34) C.F. Bokman, C. Zettersten, P.J. Sjoberg, and L. Nyholm, Anal. Chem. 76, 2017–2024 (2004).
(35) H. Hayen and U. Karst, Anal. Chem. 75, 4833–4840 (2003).
(36) S.M. van Leeuwen, H. Hayen, and U. Karst, Anal. Bioanal. Chem. 378, 917–925 (2004).
(37) H. Iwahashi and T. Ishii, J. Chromatogr. A 773, 23–31 (1997).
(38) H. Iwahashi, J. Chromatogr. B 736, 237–245 (1999).
(39) A. Baumann, W. Lohmann, B. Schubert, H. Oberacher, and U. Karst, J. Chromatogr. A 1216, 3192–8 (2009).
(40) E. Nouri-Nigjeh, H.P. Permentier, R. Bischoff, and A.P. Bruins, Anal. Chem. 83, 5519–5525 (2011).
(41) E. Nouri-Nigjeh, R. Bischoff, A.P. Bruins, and H.P. Permentier, Analyst 136, 5064–5067 (2011).
(42) W. Lohmann and U. Karst, Anal. Chem. 79, 6831–6839 (2007).
(43) U. Jurva, H.V. Wikstrom, and A.P. Bruins, Rapid Commun. Mass Spectrom. 16, 1934–1940 (2002).
(44) T. Johansson, L. Weidolf, and U. Jurva, Rapid Commun. Mass Spectrom. 21, 2323–2331 (2007).
(45) E. Nouri-Nigjeh, H.P. Permentier, R. Bischoff, and A.P. Bruins, Anal. Chem. 82, 7625–7633 (2010).
(46) E. Nouri-Nigjeh, A.P. Bruins, R. Bischoff, and H.P. Permentier, Analyst 137, 4698–4702 (2012).
(47) U. Jurva, H.V. Wikstrom, and A.P. Bruins, Rapid Commun. Mass Spectrom. 14, 529–533 (2000).
(48) U. Jurva, H.V. Wikstrom, L. Weidolf, and A.P. Bruins, Rapid Commun. Mass Spectrom. 17, 800–810 (2003).
(49) K.G. Madsen, J. Olsen, C. Skonberg, S.H. Hansen, and U. Jurva, Chem. Res. Toxicol. 20, 821–831 (2007).
(50) T. Johansson, U. Jurva, G. Gronberg, L. Weidolf, and C. Masimirembwa, Drug Metab. Dispos. 37, 571–579 (2009).
(51) W. Lohmann and U. Karst, Anal. Bioanal. Chem. 386, 1701–1708 (2006).
(52) K.G. Madsen, C. Skonberg, U. Jurva, C. Cornett, S.H. Hansen, T.N. Johansen, and J. Olsen, Chem. Res. Toxicol. 21, 1107–1119 (2008).
(53) H. Faber, D. Melles, C. Brauckmann, C.A. Wehe, K. Wentker, and U. Karst, Anal. Bioanal. Chem. 403, 345–354 (2012).
(54) T.J. Malin, L. Weidolf, N. Castagnoli, and U. Jurva, Rapid Commun. Mass Spectrom. 24, 1231–1240 (2010).
(55) K.G. Madsen, G. Gronberg, C. Skonberg, U. Jurva, S.H. Hansen, and J. Olsen, Chem. Res. Toxicol. 21, 2035–2041 (2008).
(56) K. Tahara, Y. Yano, K. Kanagawa, Y. Abe, J. Yamada, S. Iijima, M. Mochizuki, and T. Nishikawa, Chem. Pharm. Bull (Tokyo) 55, 1207–1212 (2007).
(57) K. Tahara, T. Nishikawa, Y. Hattori, S. Iijima, Y. Kouno, and Y. Abe, J. Pharm. Biomed. Anal. 50, 1030–1036 (2009).
(58) M. Lu, C. Wolff, W. Cui, and H. Chen, Anal. Bioanal. Chem. 403, 355–365 (2012).
(59) K. Nozaki, H. Kitagawa, S. Kimura, A. Kagayama, and R. Arakawa, J. Mass Spectrom. 41, 606–612 (2006).
(60) K. Nozaki, I. Osaka, H. Kawasaki, and R. Arakawa, Anal. Sci. 25, 1197–1201 (2009).
(61) U. Bussy, I. Tea, V. Ferchaud-Roucher, M. Krempf, V. Silvestre, N. Galland, D. Jacquemin, M. Andresen-Bergstrom, U. Jurva, and M. Boujtita, Anal. Chim. Acta 762, 39–46 (2013).
(62) U. Bussy, M. Delaforge, C. El-Bekkali, V. Ferchaud-Roucher, M. Krempf, I. Tea, N. Galland, D. Jacquemin, and M. Boujtita, Anal. Bioanal. Chem. 405, 6077–6085 (2013).
(63) R.G. de Lima, P.S. Bonato, and R.S. da Silva, J. Pharm. Biomed. Anal. 32, 337–343 (2003).
(64) S. Jahn, A. Baumann, J. Roscher, K. Hense, R. Zazzeroni, and U. Karst, J. Chromatogr. A 1218, 9210–9220 (2011).
(65) S. Jahn, B. Seiwert, S. Kretzing, G. Abraham, R. Regenthal, and U. Karst, Anal. Chim. Acta 756, 60–72 (2012).
(66) S. Jahn, W. Lohmann, S. Bomke, A. Baumann, and U. Karst, Anal. Bioanal. Chem. 402, 461–471 (2012).
(67) U. Jurva, A. Holmen, G. Gronberg, C. Masimirembwa, and L. Weidolf, Chem. Res. Toxicol. 21, 928–935 (2008).
(68) W. Lohmann, H. Hayen, and U. Karst, Anal. Chem. 80, 9714–9719 (2008).
(69) S. Jahn, H. Faber, R. Zazzeroni, and U. Karst, Rapid Commun. Mass Spectrom. 26, 1415–1425 (2012).
(70) S. Jahn, H. Faber, R. Zazzeroni, and U. Karst, Rapid Commun. Mass Spectrom. 26, 1453–1464 (2012).
(71) D. Melles, T. Vielhaber, A. Baumann, R. Zazzeroni, and U. Karst, J. Chromatogr. B 913–914, 106–112 (2013).
(72) T. Hoffmann, D. Hofmann, E. Klumpp, and S. Kuppers, Anal. Bioanal. Chem. 399, 1859–1868 (2011).
(73) L. Chen, D. Hofmann, E. Klumpp, X. Xiang, Y. Chen, and S. Kuppers, Chemosphere 89, 1376–1383 (2012).
(74) P.H. Gamache, D.F. Meyer, M.C. Granger, and I.N. Acworth, J. Am. Soc. Mass Spectrom. 15, 1717–1726 (2004).
(75) N.A. Mautjana, J. Estes, J.R. Eyler, and A. Brajter-Toth, Electroanalysis 20, 2501–2508 (2008).
(76) N.A. Mautjana, D.W. Looi, J.R. Eyler, and A. Brajter-Toth, Electrochim. Acta 55, 52–58 (2009).
(77) N.A. Mautjana, D.W. Looi, J.R. Eyler, and A. Brajter-Toth, Electroanalysis 22, 79–89 (2010).
(78) H. Oberacher, M. Pavlic, K. Libiseller, B. Schubert, M. Sulyok, R. Schuhmacher, E. Csaszar, and H.C. Kofeler, J. Mass Spectrom. 44, 485–493 (2009).
(79) H. Oberacher, B. Schubert, K. Libiseller, and A. Schweissgut, Anal. Chim. Acta 770, 121–131 (2013).
(80) H.P. Permentier, U. Jurva, B. Barroso, and A.P. Bruins, Rapid Commun. Mass Spectrom. 17, 1585–1592 (2003).
(81) C. McClintock, V. Kertesz, and R.L. Hettich, Anal. Chem. 80, 3304–3317 (2008).
(82) J. Roeser, H.P. Permentier, A.P. Bru ins, and R. Bischoff, Anal. Chem. 82, 7556–7565 (2010).
(83) J. Roeser, N.F.A. Alting, H.P. Permentier, A.P. Bruins, and R. Bischoff, Anal. Chem. 85, 6626–6632 (2013).
(84) Y. Zhang, Z. Yuan, H.D. Dewald, and H. Chen, Chem. Commun. 47, 4171–4173 (2011).
(85) Y. Zhang, H.D. Dewald, and H. Chen, J. Proteome Res. 10, 1293–1304 (2011).
(86) Y. Zhang, W. Cui, H. Zhang, H.D. Dewald, and H. Chen, Anal. Chem. 84, 3838–3842 (2012).
(87) T.C. Rohner, J.S. Rossier, and H.H. Girault, Electrochem. Commun. 4, 695–700 (2002).
(88) T.C. Rohner and H.H. Girault, Rapid Commun. Mass Spectrom. 19, 1183–1190 (2005).
(89) L. Dayon, C. Roussel, and H.H. Girault, J. Proteome Res. 5, 793–800 (2006).
(90) M. Prudent, J.S. Rossier, N. Lion, and H.H. Girault, Anal. Chem. 80, 2531–2538 (2008).
(91) Y. Lu, M. Prudent, L. Qiao, M.A. Mendez, and H.H. Girault, Metallomics 2, 474–479 (2010).
(92) E. Palecek, Nature 188, 656–657 (1960).
(93) G. Dryhurst and G.F. Pace, J. Electrochem. Soc. 117, 1259–1263 (1970).
(94) P. Subramanian and G. Dryhurst, J. Electroanal. Chem. 224, 137–162 (1987).
(95) R.N. Goyal, S.M. Sondhi, and A.M. Laboti, New J. Chem. 29, 587–595 (2005).
(96) A. Baumann, W. Lohmann, and U. Karst, Electroanalysis 22, 286–292 (2010).
(97) F. Pitterl, J.P. Chervet, and H. Oberacher, Anal. Bioanal. Chem. 397, 1203–1215 (2010).
(98) J.L. Ravanat, FABAD J. Pharm. Sci. 30, 100–113 (2005).
(99) R. Singh and P.B. Farmer, Carcinogenesis 27, 178–196 (2006).
(100) K.M. Li, J. Byun, M.L. Gross, D. Zamzow, R. Jankowiak, E.G. Rogan, and E.L. Cavalieri, Chem. Res. Toxicol. 12, 749–757 (1999).
(101) D.W. Looi, J.R. Eyler, and A. Brajter-Toth, Electrochim. Acta 56, 2633–2640 (2011).
(102) S. Plattner, R. Erb, F. Pitterl, H.J. Brouwer, and H. Oberacher, J. Chromatogr. B 883–884, 198–204 (2012).
Herbert Oberacher and Florian Pitterl are with the Institute of Legal Medicine and Core Facility Metabolomics at Innsbruck Medical University in Innsbruck, Austria. Direct correspondence to: herbert.oberacher@i-med.ac.at














Best of the Week: AI and IoT for Pollution Monitoring, High Speed Laser MS
April 25th 2025Top articles published this week include a preview of our upcoming content series for National Space Day, a news story about air quality monitoring, and an announcement from Metrohm about their new Midwest office.




LIBS Illuminates the Hidden Health Risks of Indoor Welding and Soldering
April 23rd 2025A new dual-spectroscopy approach reveals real-time pollution threats in indoor workspaces. Chinese researchers have pioneered the use of laser-induced breakdown spectroscopy (LIBS) and aerosol mass spectrometry to uncover and monitor harmful heavy metal and dust emissions from soldering and welding in real-time. These complementary tools offer a fast, accurate means to evaluate air quality threats in industrial and indoor environments—where people spend most of their time.


