On-line and Off-line 2-D LC–ESI MS-MS Methods in Proteomic Analysis
Special Issues
Off-line 2-D LC–MS-MS represents a powerful alternative to on-line methodologies for protein identification from complex proteomes, improving the chromatographic resolution of digest peptide mixtures, even for low-abundance proteins. Here, the authors provide a detailed comparison of the two techniques.
Increasingly, multidimensional, and in particular two-dimensional liquid chromatography (2-D LC) is becoming an important alternative to 2-D gel electrophoresis (2-D GE) in the separation of certain classes of protein digests for analysis by mass spectrometry and tandem mass spectrometry (MS and MS-MS, respectively). Despite the fact that 2-D GE is capable of addressing samples containing upwards of 2000 proteins across a six-fold concentration range, this technique is limited in the analysis of low-abundance/low-molecular-weight proteins, as well as acidic, basic, hydrophobic and high-molecular-weight proteins. Among this subset of the proteome are signal receptors and transducers, transcription controllers and other regulatory and signaling proteins of interest in the investigation of cellular-level disease pathways. In addition to losses resulting from manual manipulation and transfers of sample, analyte recovery by 2-D GE is hindered by the rapid diffusion of low-abundance analytes out of the gel spot prior to extraction and the obscuring of low-abundance analytes by electrophoretically co-migrating higher-abundance proteins. Furthermore, 2-D GE is difficult to automate and does not couple readily to the mass spectrometer. As a result, employment of the technique can be tedious and time-consuming and could lack adequate reproducibility.


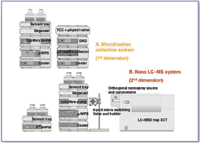
Figure 1. A two-dimensional LC-MS instrument system. Both on- and off-line configurations comprise a nanoflow pump with a micro vacuum degasser; thermostatted micro well-plate autosampler; two-position/six-port micro switching valve box with holder; SCX ion-exchange column (first-dimension); RPLC analytical and enrichment nanocolumns (second-dimension) with matching low dead volume tubing, fitting, and holdings; a quarternary pump and LC-MSD Trap XCT with an orthogonal nanospray source. Off-line configuration (shown here): An Agilent 1100 Series Micro-Fraction Collection System comprising a capillary pump with a micro vacuum degasser, thermostatted well-plate autosampler, thermostatted column compartment with a micro two-position/six-port micro valve, diode-array detector with 500 nL cell, and thermostatted microfraction collector is interposed between the SCX and RPLC separation dimensions.
Two-dimensional LC eliminates the methodological deficiencies encountered in separating low-abundance/low-molecular-weight proteins and other protein analytes that are difficult to resolve by 2-D GE. By linking two orthogonal chromatographic separation mechanisms, such as strong cation exchange (SCX) or strong anion exchange (SAX) with reverse-phase liquid chromatography (RPLC), 2-D LC can achieve sensitivities comparable or superior to 2-D GE. (The overall resolving power of a multidimensional chromatographic method employing orthogonal separation mechanisms approximates the product of the resolution or peak capacity of each phase (1). The order of the chromatographic phases is dictated, in part, by the need to desalt the SCX eluent prior to carrying out RPLC chromatography. It also serves to properly condition the eluent stream from a large excess of salt ions that could negatively impact analyte ionization and confound the MS analysis. Two-dimensional LC methods have the inherent flexibility of LC to tailor both stationary and mobile phases to match sample characteristics in a way that maximizes resolution and recovery. All 2-D LC operations are automated, thereby avoiding manual handling losses. Sample size limitations can be compensated for, in part, by in-line concentration and by the use of an appropriate gradient that provides sufficient chromatographic resolution to avoid detection problems associated with co-eluting peaks. Even where there is co-elution, electrospray ionization (ESI), when used in conjunction with 2-D LC, produces a rich complement of peptide ions and fragmentation products that enhance the likelihood of detecting otherwise obscured low-abundance analytes. This is particularly relevant when MS detection sensitivities can be augmented by the ion selection and concentration capabilities of ion trap MS.


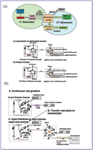
Figure 2. a) An on-line 2-D LC proteomic analysis workflow. In a typical on-line 2-D LC workflow, a mixture of tryptic peptides are eluted from an SCX column using stepwise isocratic injections of an aqueous salt of increasing concentration. After each injection, the analytes are captured on the C18 enrichment column, washed free of salt and, after a valve switch, back flushed onto the C18 analytical column and chromatographed using an AcN/H2O gradient with increasing organic content. The eluent from the analytical column then is sprayed directly into the mass spectrometer. b) An off-line 2-D LC proteomic analysis workflow. In the off-line workflow, the SCX column is eluted using a linear salt gradient, and equally timed microliter fractions are collected in plate wells using the microfraction collector. After any intervening operations, each fraction, in turn, is injected onto the second-dimension C18 enrichment column by means of an autosampler, and further operations are allowed to proceed in a manner identical to the on-line workflow.
Instrument Platform and Flow Control
Figure 1 depicts the components of a typical instrument system that can be employed for both on-line and off-line 2-D LC. To integrate the complex arrangement of LC flow pathways within a reasonably compact footprint, these instruments utilize programmable systems of valves to switch between chromatographic dimensions, as well as between the various phases of a particular separation (e.g., column equilibration, back flushing, column loading, chromatography, etc.). At nanoscale volumes, it is crucial that these valves (as well as other flow containing components and connections of the system) have the smallest possible internal volumes so as to minimize peak broadening. This constraint is critical when analytes of interest are present at concentrations near or at the method's limit of detection and peak broadening could cause analyte concentrations to fall below the detection threshold. Figures 2a and 2b illustrate an off-line and an on-line LC system, respectively, that incorporates two two-position/six-port microvalves to control system flows.



Table I. On-line and Off-line Chromatographic Method Procedures
Precise flow control is another important requirement for optimizing the resolution, sensitivity and reproducibility of nanoscale LC–MS performance. Many capillary LC systems depend upon a passive splitter with a preset split ratio to control flow rate. As the scale of LC operations and the associated flow rates are downsized, this approach becomes increasingly inadequate for maintaining constant flow within acceptable limits. At the nanoscale, even small perturbations in flow caused by variations in pump performance, changes in gradient mobile-phase compressibility or a flow-path obstruction can result in a loss of resolution that precludes good peak separation or causes specific peak intensities to fall below the threshold limit, thereby going undetected. An approach employed by Agilent Technologies (Palo Alto, CA) to remedy this problem involves the use of nanoflow chromatographic pumps equipped with an active feedback electronic flow control (EFC) system that monitors and compensates for flow-rate deviations. The electronic flow control consists of a flow sensor and an electromagnetic proportioning valve (EMPV). The flow sensor measures the flow rate and controls the active splitting between a waste flow and a column flow with the EMPV. This concept makes the column flow independent from back-pressure and delivers stable and reliable nanoflow rates.



Table I. On-line and Off-line Chromatographic Method Procedures (continued)
Differentiating On- and Off-line 2-D LC
In on-line 2-D LC, digest peptides bound to the SCX (or SAX) column are eluted using an autosampler programmed for the stepwise injection of specific volumes of salt solution of increasing concentrations. After each injection, the eluted peptides are placed on a short trapping column, washed free of salt and then back- flushed onto an analytical nanobore column and chromatographed. Each discrete step is accompanied by a programmed valve switch in order to configure the flow path for the appropriate stage of the analysis. On-line methods are more completely automated than the corresponding off-line approach and require less of an equipment investment. Therefore, they are more cost-effective for analyzing simple to moderately complex peptide digest mixtures.



Figure 3. Precision collection principle for lower õL range volumes. During fraction collection, the capillary tip of the microfraction collector moves continuously upwards, keeping the forming droplet in contact with the surface of the deposited liquid until the well is filled. When fraction collection is complete, the capillary tip moves quickly upwards, ensuring that the final droplet is delivered to the well. The well-filling velocity is calculated from both the actual flow rate and the geometry of the well. This collection process ensures precise, bubble-free delivery of liquid into a variety of well types and vessels and prevents cross-contamination between collected fractions.
The off-line method exhibits its greatest utility in the analysis of highly complex peptide digest mixtures. Instead of stepwise isocratic injections of increasing salt concentration, the off-line method's first-dimension chromatography employs a salt gradient to elute and collect peptide analytes at specified intervals. The use of a gradient in the first dimension significantly improves peak capacity, as well as the overall resolution profile, while shortening the run time. The upstream resolution improvement impacts downstream separation and detection, producing a richer suite of data and, therefore, a more robust protein identification. Moreover, providing an off-line interval between the first and second chromatographic dimensions gives the method considerable flexibility for additional sample modification or tagging between separations.



Figure 4. MS-MS spectrum of the peptide from orothate phosphoribosyltransferase. (Protein number 140 in the off-line experiment, score 9.28, peptides 1, spectra 1, sequence coverage 8%, MW 24.6 kDa, pI 5.80.) The fragmentation pattern provides sufficient data for conclusive identification of the protein. This is one of the 33 proteins found only in the off-line method.
Fraction Collection
Implementation of the off-line method requires that the LC platform be equipped with a programmable fraction collection system capable of precisely and reproducibly dividing the SCX eluent into timed small volume aliquots. A micro-fraction collector is used for this purpose (2). It enables fraction collection based upon peak detection or time slices and is equipped with Peltier cooling to prevent thermal decomposition and evaporation of microscale fractions — an important consideration with very small volumes. To ensure reproducible collection, a new dispensing principle is utilized: the tip of the capillary moves upward continuously so that it remains in contact with the liquid surface as each well fills. This enables a sharp cutoff of the dispensing process and prevents both the formation of air bubbles in the collection wells and crosscontamination between successive fractions. (Figure 3).



Table II. Comparison of the highest protein scores for on-line and off-line 2-D LC-MS analysis of the yeast cell lysate.
Sample Preparation
Lyophilized yeast cells (Saccharomyces cerevisiae), resuspended in cooled 50 mM NH
4
HCO
3
containing 8 M urea, were disrupted in a bead beater with 0.5 mm glass beads (BioSpec Products, Bartelsville, OK). After centrifugation to remove cell debris, proteins in the supernatant were reduced with 1 mM DTT at 37 °C for 1h, alkylated in the dark with 10 mM iodoacetamide for 30 min at room temperature, ultrafiltrated for buffer exchange and tryptically digested with TPCK trypsin at 37 °C for 16 h. Finally, the sample was lyophilized in a SpeedVac (Bachofer, Reutlingen, Germany) and dissolved in 5% AcN, 0.03% formic acid prior to analysis.


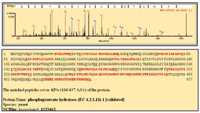
Figure 5. MS-MS spectrum of one identified peptide from phosphopyruvate hydratase. The specific peptide sequence, as well as the total sequence coverage and the database accession number of the identified protein, is shown below the spectrum.
Application
To compare the results achieved by on- and off-line 2-D LC, identical samples of trypsin-digested yeast (Saccharomyces cerevisiae) proteome were subjected to both off-line and on-line 2-D LC, followed by MS and MS-MS analysis. The same conditions, solvents and columns were used in each case. In the on-line version, injections of a series of isocratic aqueous salt solutions at stepped concentrations were adjusted to approximate comparable fractions collected in the off-line approach. The sample preparation is described earlier, and Table I documents the on-line and off-line LC and MS method protocols (3).


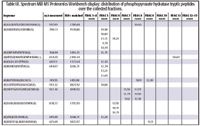
Table III. Spectrum Mill MS Proteomics Workbench display: distribution of phosphopyruvate hydratase tryptic peptides over the collected fractions.
Data Analysis and Management
MS-MS data acquired for each method (4) were evaluated using the Spectrum Mill MS Proteomics Workbench (Agilent Technologies). Three replicate analyses produced identical results: the number of proteins identified in the on-line and off-line method was 101 and 144, respectively. Typically, identification scores for the same proteins are significantly higher in the off-line analysis. This result indicates that the salt gradient in the off-line method yields better resolution compared with the isocratic stepped series of injections of salt solutions of increasing concentration used to elute the SCX column in the on-line method. The improvement in resolution has repercussions on the downstream MS-MS analysis, producing greater sequence coverage and, therefore, higher scores in cases where the same proteins are identified by both methods (Table II). In addition, the resolution improvement of the off-line method enables the detection of low-abundance peptides corresponding to the 43 additional proteins that go undetected by the on-line method. This is demonstrated in Figure 4, in which the MS-MS spectra for one peptide provide conclusive data for the identification of the protein orothate phosphoribosyltransferase.
One of the challenges in proteomics research is data management. Software tools are required to collect and organize the large amounts of data scattered across multiple LC fractions and the MS-MS data extracted from them. There also is a need to efficiently organize and compare cross-platform data and data across multiple experiments. This type of data assembly not only forms the basis for automated database searching and/or de novo sequencing but also helps to extract patterns in the data that reveal cellular processes where collections of proteins interact. Table III illustrates this type of data organization, showing results for the tryptic peptides derived from the protein phosphopyruvate hydratase (identified in the off-line experiment), which are distributed over the collected fractions. The fact that the majority of the relevant digest peptides are confined to a single fraction further demonstrates the resolution enhancement achieved by the gradient SCX separation employed in the off-line method. Figure 5 shows an MS-MS spectrum for one of the phosphopyruvate hydratase peptides. The suite of y- and b-series product ions are indicated. The lower panel details the database search results, indicating all identified peptides, as well as protein identity, protein sequence, sequence coverage, the protein species and its database accession number.
Conclusion
Off-line 2-D LC–MS-MS represents a powerful alternative to on-line methodologies for protein identification from complex proteomes. The results shown here demonstrate that a significantly greater number of proteins (approximately 40%) are identified than in a comparable on-line 2-D LC–MS analysis. Moreover, even for proteins detected by both approaches, the off-line identifications are more robust. In addition to yielding a greater complement of information, the higher confidence of the identifications reduces the probability of identifications that are false positives.
Because the overall improvement in results is attributed to the resolution enhancement from the use of gradient chromatography in the first dimension, it is anticipated that further gains in this direction could be made by performing the SCX chromatography with an even longer and shallower gradient mobile phase. However, this would require the collection of additional fractions and would significantly increase analysis time. Thus, one has to consider the tradeoff between the amount of data to be gained and overall analytical throughput. Also, while the number of proteins identified would increase under these more intensive separation constraints, so too would the number of false positives, so practical experience is required to properly set the methodological parameters.
In addition to data enhancement, the off-line methodology provides a great deal more flexibility in carrying out a multidimensional chromatographic separation. Other separation or prefractionation techniques can be incorporated into the method as well. For convenience, fractions can be stored for later or repeated analysis, allowing more flexibility in distributing the analysis workload or in utilizing the equipment. For those performing screening or other studies, easy access to the fractionated peptides at an intermediate stage facilitates a variety of assessments, as well as additional sample processing — such as chemical or enzymatic modification — prior to the resumption of the second dimension chromatography and subsequent MS and MS-MS analysis.
References
1. T. Wehr,
LCGC
20
(10), 962 (2002).
2. Agilent Technologies. Agilent Micro-Fraction Collection System, Product Brochure, Publication Number 5988-9614EN, 2003.
3. R. Moritz, M. Vollmer and E. Nägele, Improved protein identification: Off-line multidimensional LC-MS as an effective tool for proteomics research, Agilent Publication Number 5988-9913EN, 2003 and literature cited therein.
4. Agilent Technologies. Agilent Spectrum Mill MS Proteomics Workbench, Product Brochure, Publication Number 5988-9443EN, 2003.
Martin Vollmer is an R&D biochemist and Edgar Nägele is an LC–MS applications chemist at Agilent Technologies R&D and Marketing GmbH & Co., KG. E-mail: edgar_naegele@agilent.com Ralf Moritz is with Sandoz GmbH in Kundl, Austria.

















Mass Spectrometry for Forensic Analysis: An Interview with Glen Jackson
November 27th 2024As part of “The Future of Forensic Analysis” content series, Spectroscopy sat down with Glen P. Jackson of West Virginia University to talk about the historical development of mass spectrometry in forensic analysis.

Detecting Cancer Biomarkers in Canines: An Interview with Landulfo Silveira Jr.
November 5th 2024Spectroscopy sat down with Landulfo Silveira Jr. of Universidade Anhembi Morumbi-UAM and Center for Innovation, Technology and Education-CITÉ (São Paulo, Brazil) to talk about his team’s latest research using Raman spectroscopy to detect biomarkers of cancer in canine sera.



