Mass Spectrometry Forum - Mass Calibration: Cluster Calibrants for Higher Masses
Part III of this four-part series continues the discussion of mass calibration.
Part II of this four-part series described the use of perfluorinated compounds as calibrants for electron ionization (EI) and chemical ionization (CI) mass spectrometry (MS), and described links between time/intensity files recorded by a data system, the calibration file, and the properly calibrated mass spectrum (1). Here, Part III describes calibrants that are used in higher masses accessed with other ionization methods. Many of the basic details of calibration remain the same, including the linkage between time/intensity files and the calibration file, and the fact that an external calibration (using a calibrant separately introduced into the ionization source of the mass spectrometer) is intrinsic to proper operation of the mass spectrometer.



Kenneth L. Busch was among those curious about the origin of the chemical noise âÂÂpeak at every massâ in a FAB mass spectrum, musings that ultimately led to his development of charge-transfer derivatization in MS. Views expressed in this column are those of the author and not those of the National Science Foundation. Contact the author at: wyverners@yahoo.com.
Electron ionization and chemical ionization are used for sample compounds from which a population of stable gas-phase molecules can be created through evaporation at (usually) elevated temperatures. Because sample volatility generally is inversely proportional to molecular mass (other factors also contribute), the mass range concordant with the use of EI and CI sources tops out at perhaps 1500 Da. With higher mass, and especially for most biomolecules, thermal stability is low, and direct evaporation of such compounds at higher temperatures cannot be achieved without decomposition. Accordingly, ionization methods other than EI and CI must be used to produce ions with higher masses extending to several thousand daltons. Details of the calibration process will change as the ionization method changes, as the breadth of the calibrated mass range increases, and as the type and use of the mass spectral data change. Figure 1 illustrates this latter point, plotting the fraction of the mass range from which analytical data typically is drawn against molecular mass. The situation is illustrated in the figure for a single stage of mass analysis; for tandem MS (MS-MS) analyses, the relationship differs.


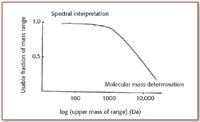
Figure 1. Generalized relationship between the fraction of the mass spectrum from which analytical information is drawn and the molecular mass of the compounds investigated.
The perfluorinated calibrant FC-43 used for EI calibration contains in its mass spectrum an ion at m/z 614, representing the high end of the useful mass range. As described in Part II, other perfluorinated compounds can be used to extend the calibration to still higher masses; these other calibrants have molecular masses higher than that of FC-43. Their use is predicated on the hope that the higher mass molecular ion and fragment ions could still be produced with discernible relative abundances. But as the molecular mass of the calibrant increases, the compound volatility decreases, and the relative abundances of the molecular ion and high mass fragment ions usually decreases. A strategy to reach higher calibrated mass ranges by increasing the molecular mass of the calibrant compound soon becomes inadequate.
Using ionization methods such as liquid secondary ion mass spectrometry (LSIMS) and fast atom bombardment (FAB), and (in common usage today) electrospray ionization (ESI) and matrix-assisted laser desorption ionization (MALDI), the zenith of the mass range extends routinely to several thousand daltons (kDa), intermittently to several tens of thousand daltons, and exceptionally approaches the megadalton (MDa) mark. Calibration needs certainly change as analyses extend to these higher masses. Discussion here follows the chronological history of mass calibrants developed first for FAB and LSIMS, and then extends to calibrants developed (and still developing) for ESI and MALDI. Basic changes in strategies for instrument calibration itself (for example, no longer using an external calibration), the analytical subtleties of the higher mass territories, and missteps taken along the path to higher masses, are reserved for discussion in Part IV of this series.
High mass calibrant compounds form higher mass ions through a process of clustering. Represented in a simple nomenclature, a calibrant compound of molecular mass M forms cluster ions Mn where n ranges from 1 to 50, and in which the charge can be either positive or negative. Ionic clusters consisting of cations (C) and anions (A), with a single ion excess of either the cation or the anion, also are used. Simply, and as reasonably expected, the relative abundances of cluster ions tend to decrease with higher mass, with some interesting exceptions for cluster ions of specific structures and exceptional stabilities. Mass intervals between ions of useful abundance in the mass spectrum of the calibrant compound are determined by the molecular mass of the calibrant in the case of a molecular cluster Mn, or the mass of the CA unit in an ionic cluster. The desirability of using a calibration compound composed of monoisotopic elements remains, because as before, this characteristic maximizes the single-peak relative abundances of each of the calibrant ions. With the use of a clustering calibrant, however, rather than controlling the dissociative processes of a molecular ion, rational selection instead focuses on the tendency for the cluster ion to "stick together," based upon non-covalent interactions, or cemented with a singly charged charge carrier such as a proton, a halide ion, or a single excess cation or anion derived from the calibrant itself. Ideally, each molecular or ionic subunit should be robust and resistant to internal fragmentation, because these dissociative processes would detract from the relative abundances of the cluster ion. These desiderata are summarized in Figure 2.



Figure 2. Generalized scheme for desirable characteristics of a clustering calibrant compound.
The first applications of FAB/LSIMS in the 1980s, showcasing the newfound ability to ionize nonvolatile sample compounds with kilodalton masses, presented an immediate challenge to the standard procedures for instrument calibration. Assuming that the instrument response was stable, analysts extended the bar to masses much higher than the highest mass ion created from the calibrant compound. More importantly, external calibrants of the usual sort did not produce a mass spectrum with these new ionization methods. For a short time, the chemical noise that is a characteristic of the FAB mass spectrum (reflecting the sputter-induced chemistry of the liquid support matrix) was a blessing in disguise, as the masses of ions from the sample were established by manual count starting from ions of known masses. Such a mass spectrum was not mass calibrated externally through an instrument calibration then, but was internally calibrated. The origins of these near-baseline ions occupied the attentions of the chemically curious for some time, but their more immediate usefulness for mass counting is evident. Given an established consensus mass of any one ion in the mass spectrum and the resolved peaks at every integral mass, the integral masses of higher mass ions could be established by counting. Uncertainties in the mass assignments deduced in such fashion increased as the counting proceeded to higher masses, and as baseline peaks appeared with lower intensities. As we have described in previous columns, the isotopic envelope of higher mass ions begins to spread out over several integral masses. This is true of the background ions formed for a glycerol support matrix as it is for ions from sample molecules. Clearly, the practice of counting every background ion mass in a FAB or LSIMS mass spectrum to determine the masses of the sample-related peaks would not long suffice.
What external calibrant compound might then be chosen for use in FAB and LSIMS? How might a calibrant be chosen so that calibrant ions appear with reasonable relative abundances at regular intervals within a wide mass range, and not be dependent upon evanescent background signals? The first practicable answer was serendipitously apparent in the liquid matrix used to dissolve and support the analyte molecules for FAB and LSIMS ionization. Glycerol (G), for example, forms a series of cluster ions in the positive ion mass spectrum GnH+. The protonated molecule of glycerol (n = 1) would then appear at m/z 93, and the protonated dimer (n = 2) at m/z 185, and the protonated trimer at m/z 277, and so on, with decreasing relative abundances for higher mass cluster ions. For negative ions, analogous cluster ions (Gn-H)- are observed (see Figure 3). Glycerol cluster ions with n up to 15 and 20 could be observed at high detector gains, and the masses of these ions were, of course, predictable and precisely known. This clustering of the glycerol molecules, each cluster associated with a single proton, or deficient in a single proton, provided a means to calibrate the mass spectrum up to a mass of perhaps 2000 Da, with the higher intensity calibrant ions separated by the mass of glycerol itself (92 Da). Use of the glycerol clusters combined an internal and an external mass calibration process. A sample of pure glycerol could be used to create a separate external instrumental calibration file. The mass spectrum recorded from the sample itself would also contain ions corresponding to these glycerol clusters, and their appearance at the correct masses provides an internal check of consistency.


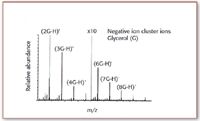
Figure 3. Negatively charged cluster ions in the FAB mass spectrum of glycerol (G).
Glycerol soon was augmented via exploration of the practical utility of many other liquid matrices; hundreds were explored eventually before common practice settled on a small number of generally suitable liquid matrices for sample preparation in FAB/LSIMS (2). Other support matrices also formed cluster ions of masses higher than the molecular mass that could be used for calibration. Soon, however, the limitations of using cluster ions from the matrix became evident, both as a practical matter in terms of the mass range but also the need to maintain an instrumental calibration separate from the matrix used for sample preparation, mass interferences that needed to be avoided, and the simple fact that an ideal support matrix would be itself transparent in the recorded mass spectrum. A change in the basic approach to calibration to embrace clustering calibrant compounds became apparent with the first use of the cluster ions generated from the liquid support matrix. Once this path was chosen, other potential calibrant compounds that produced cluster ions in their sputtered mass spectra were investigated, with a careful eye towards maximizing the intensity of the cluster ions, minimizing the width of the isotopic envelope for higher mass ions, and maintaining a separate external calibration process. Alkali halide salts (and mixtures of them) were established to provide cluster ions in both positive and negative ion FAB/LSIMS that could be used for calibration. The positive ion cluster ions Cn+1An+ contained a single excess cation (C) that provided the single positive charge for the cluster; the negative ion cluster ions CnAn+1- contained a single excess anion (A). Cesium iodide is an especially useful calibrant compound. The integral mass of the cesium cation is 133 Da, and that of iodide anion is 127 Da, resulting in calibration ions that are spaced reasonably in both the positive and the negative ion mass spectra. Furthermore, each of these elements is monoisotopic, maximizing the intensity of the calibrant ion. Using cesium iodide as the calibrant, reliable mass calibration could be extended to 5 kDa and, with a little added effort, to 10 kDa. For more closely spaced calibrant ions, a mixture of CsI, RbI, and NaI also can be used, providing a wider variety of calibrant ions that contain mixtures of the three different cations. Cluster ions formed from cesium iodide have been observed to masses of up to 20 kDa, with an (almost) smoothly decreasing intensity curve (3–5). Deviations of observed ion intensities from the expected smooth abundance decay curve led to a brief flurry of research into "magic numbers" of ions in the gas phase; this topic is worthy of a separate future discussion. Calibration tables with the masses of calibrant ions from cesium iodide and other alkali halide mixtures are found in appendices of several mass spectrometry textbooks, and are available readily from manufacturers and from web resources. Such calibration tables also are preloaded into instrument software control systems.
Once the strategy of exploring cluster ions for calibration to higher mass ranges became established, other potential calibrants were explored and described in the literature. Blaum et al. (6) described the use of atomic gold clusters for calibration; cluster ions of the form Aun- with n up to 50 provided a calibration to 10 kDa. Letzel et al. (7) describe an organic compound that provides stable cluster ions in both the negative and position ion modes of electrospray ionization, with the consequential suggestion that these ions could be used to provide mass calibration to a mass limit of 6 kDa. Konig and Fales (8) described organic salts of cesium that could be used for calibration in ESI up to a high mass limit of 10 kDa. Research into other calibrant compounds continues; the use of cesium iodide itself has the advantage of being simple and direct.
As we foresee topics covered in the final Part IV of this series on calibration in MS, several issues become clear that complicate the "simple and direct" approach exemplified by cesium iodide. The first issue is a fact apparent in practice, and implicit in the simple relationship evident in Figure 1. Attempts to increase the breadth of the calibrated mass range are inconsistent with the fact that, in practice, the entire mass spectrum was not usually of interest. For example, a useable calibrated range of perhaps 2 kDa could be placed at many different places in the range, viz 1–3 kDa, 5–7 kDa, or 18–20 kDa. Seldom was a single mass spectrum from 0–20 kDa needed, so seldom is such a mass spectrum recorded or interpreted. For higher resolution mass measurements, even smaller relevant mass ranges were established. To coin a phrase, "concordant calibration" matches the calibrated mass range to the mass range from which the relevant analytical information is to be recorded. Selection of a concordant calibrant is part of intelligent experimental design.
Secondly, in appreciation of basic analytical metrology, we note that calibration of mass spectrometers using either an external or internal calibration relies on linkage to the absolute mass scale based upon 12C. Cluster ions based upon 12C can be created with any of several ionization methods, and such cluster ions can also be used for direct calibration of the mass spectrometer. We cover these and other topics in calibration in Part IV of this series.
References
1. K.L. Busch, Spectroscopy 20(2), 76 (2005).
2. E. DePauw, Mass Spectrom. Rev. 5, 191–212 (1986).
3. I. Katakuse, H. Nakabushi, T. Ichihara, T. Sakurai, T. Matsuo, and H. Matsuda, Int. J. Mass Spectrom. Ion Proc. 57, 239–242 (1984).
4. M. E. Castro and D. H. Russell, Anal. Chem. 57, 2290–2293 (1985).
5. Y. J. Twu, C. W. S. Conover, Y. A. Yang, and E. A. Bloomfield, Phys. Rev. B 42, 5306–5316 (1990).
6. K. Blaum, G. Huber, H.-J. Kluge, and L. Schweikhard, Eur. J. Phys. D 24, 145–148 (2003).
7. M.C. Letzel, C. Agena, and J. Mattay, Eur. J. Mass Spectrom., 7, 35–38 (2001).
8. S. Konig and H.M. Fales, J. Am. Soc. Mass Spectrom.10, 273–276 (1999).












Mass Spectrometry for Forensic Analysis: An Interview with Glen Jackson
November 27th 2024As part of “The Future of Forensic Analysis” content series, Spectroscopy sat down with Glen P. Jackson of West Virginia University to talk about the historical development of mass spectrometry in forensic analysis.

Detecting Cancer Biomarkers in Canines: An Interview with Landulfo Silveira Jr.
November 5th 2024Spectroscopy sat down with Landulfo Silveira Jr. of Universidade Anhembi Morumbi-UAM and Center for Innovation, Technology and Education-CITÉ (São Paulo, Brazil) to talk about his team’s latest research using Raman spectroscopy to detect biomarkers of cancer in canine sera.



