Mass Spectrometry Techniques to Unravel the Heterogeneity of Glycoproteins
Special Issues
Since glycans are responsible for bioactivity, solubility, immunogenicity, and clearance rate from circulation, it is vital to have a detailed map of glycans in therapeutic glycoproteins. Detailed glycoprotein structural analysis must be able to identify the peptide sequence where the glycans are attached as well as the structure of the glycan portion, including oligosaccharide sequence and glycosyl linkages. This article details methods for mass spectrometry experiments on both released glycans (“glycomics”), as well as on intact glycopeptides (“glycoproteomics”) using electron transfer dissociation, high-energy collision dissociation, and collisioninduced dissociation fragmentation pathways, which are needed to fully elucidate the structure of glycoproteins.
The production of high-quality pharmaceutical recombinant therapeutic glycoproteins with consistency in glycan quality is still challenging. Since glycans are responsible for bioactivity, solubility, immunogenicity, and clearance rate from circulation, it is vital to have a detailed map of glycans in therapeutic glycoproteins. Detailed glycoprotein structural analysis must be able to identify the peptide sequence where the glycans are attached as well as the structure of the glycan portion, including oligosaccharide sequence and glycosyl linkages. We detail methods for mass spectrometry (MS) experiments on both released glycans (“glycomics”), as well as on intact glycopeptides (“glycoproteomics”) using electron transfer dissociation (ETD), high-energy collision dissociation (HCD), and collision-induced dissociation (CID) fragmentation pathways, which are needed to fully elucidate the structure of glycoproteins. We also show additional protocols of a combination of glycosyl composition and glycosyl linkage analysis, using a combination of methylation analysis, multiple-stage mass spectrometry (MSn), and exoglycosidase digestion, and provide information on the glycan topology as well as detection methods for potential nonhuman modifications that could arise from mammalian expression systems such as Galα1-3Gal and N-glycolylneuraminic acid (NeuGc). Our consolidated experiments outline all the necessary information pertaining to the glycoprotein, including glycan fine structure, attachment site, and glycosylation degree to be obtained for pharmaceutical recombinant glycoproteins.
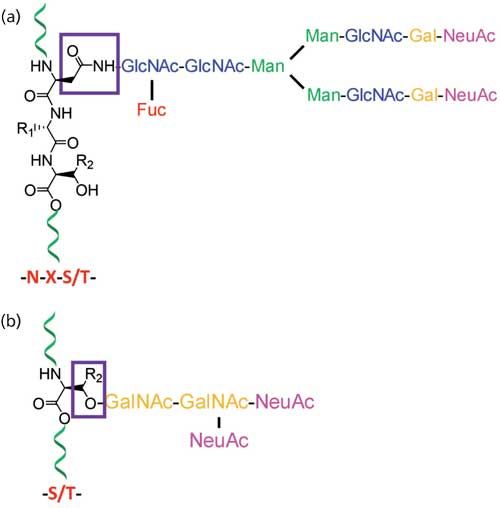
At least half of all proteins in living organisms are glycosylated, so the importance of structural characterization of glycoproteins is increasing rapidly (1). Glycans directly or indirectly influence many cellular physiological functions, and the study of precise glycan structure, their structural variability, their sites of attachment to the protein, and the degree to which these sites are occupied are vital in deducing their functional roles. Nontemplate-driven biosynthesis and microheterogeneity of glycosylation often make the structural assignment tasks difficult (2). A set of glycosyltransferases drives the biosynthesis of glycans, and glycans on a glycosylation site exist as mixtures of heterogeneous structures. Structural determination of carbohydrates from complex biological samples are most commonly determined by analytical techniques using electrospray ionization mass spectrometry (ESI-MS), matrix-assisted laser desorption-ionization MS (MALDI-MS), capillary electrophoresis (CE), and nuclear magnetic resonance (NMR) spectroscopy (3). The composition of monosaccharides in glycans and their branching is determined by MS, whereas gas chromatography (GC) (after chemical derivatization) or NMR is used for the determination of linkage information and monosaccharide types. These complementary techniques are often required to be used together for the comprehensive determination of glycosylation (4,5). MS is one of the major techniques for the analysis of glycoproteins that are usually available as a heterogenic mixture in minute amounts since it can, in principle, be used for the analysis of complex mixtures of samples of low abundance. Mammalian glycans, which consist of a limited assortment of monosaccharides, are often isomeric, having the same molecular mass. The glycans are usually multiply branched and exist as mixtures of various branching and substitution patterns (6). Since numerous potential attachment points exist in each monosaccharide, multitudes of isomeric structures are possible. Stereoisomers such as mannose and galactose, which are not quite distinct based on their molecular mass, produce a slightly different ring cleavage pattern in tandem multiple-stage mass spectrometry (MSn) analysis. Thus, analytical techniques such as glycosyl linkage or composition analysis by GC–MS of partially methylated alditol acetates (PMAAs) generated from glycans are performed to distinguish stereoisomers (7). Glycans that are found on the cells in the form of either glycoproteins or glycolipids are covalently linked to proteins or lipids, respectively. The linkages between two monosaccharides are called glycosidic bonds and the linkages between glycans and proteins are classified as either N-linked or O-linked. In N-linked glycans, the linkage between glycan and protein is through the side chain nitrogen of asparagine. On the other hand, glycans involving linkage through the side-chain oxygen of serine or threonine of peptides are O-linked glycans (Figure 1) (6).


Figure 1: Typical structure of protein glycosylations that are classified into (a) N-linked glycans where glycans are attached to the side chain of asparagines (N) on N-glycan consensus sequence NXS/T; and (b) O-linked glycans where glycans are attached to either serines or threonines of proteins.
MALDI-time-of-flight (TOF) MS is one of the most common techniques used for glycan characterization, and it enables rapid and sensitive analyses of singly charged larger biomolecules (8). Because of the structural complexity and low ionization efficiency of carbohydrates that results from their hydrophilicity, MALDI-TOF MS analyses are usually performed after the permethylation of glycans, which improves their sensitivity for MS detection by increasing the ionization efficiency of glycans up to 20-fold (9). Other complementary techniques such as ESI-MS and MSn fragmentation enable further structural characterization of selected glycan ions, and that helps in the differentiation of “isobaric” glycans, which have the same mass but different sugar compositions, linkages, or structures (10). Liquid chromatography–tandem mass spectrometry (LC–MS/MS) analysis of permethylated glycans assists in obtaining the structural determination of isomers and further selective fragmentation. Recently, better chromatographic separation and trapped ion mobility spectrometry (TIMS) was used for the characterization of isomeric glycans (10,11).
This article is intended to provide a brief overview of the general techniques involved in the characterization of heterogeneous glycoproteins, such as the determination of glycan structure, sites of glycosylation, site-specific glycan heterogeneity, and glycosylation site occupancy of glycoproteins using MS.
Determination of Protein Glycosylation by MS
Comprehensive analysis of the cellular glycan repertoire is essential for the study of underlying mechanisms in complex biological processes such as intra- and intercellular signaling, organ development, immunological responses, tumor growth, and even stability of bioconjugates. When the structural analysis of protein glycosylation is performed with the released glycans, the approach is termed glycomics. On the other hand, the analysis of glycosylation on proteins without its release is termed as glycoproteomics (Figure 2). The most common analytical procedures for the characterization of glycosylation comprise the analysis of the individual glycan structures in detail along with their isomeric pattern (glycomics) and detailed evaluation of site of glycosylation on glycoproteins and glycopeptide characterization (glycoproteomics), including the glycan variability and degree of occupancy of the site (6,12).


Figure 2: MS-based techniques used to determine the N- and O-linked glycosylation (PTM) on glycoproteins by glycomics and glycoproteomics.
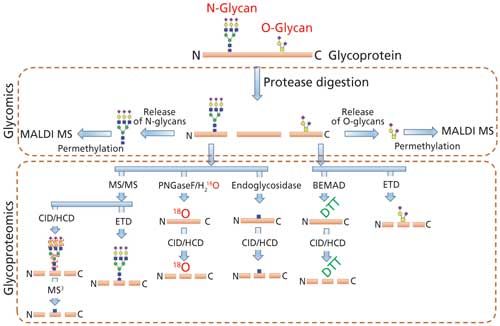
The analysis of glycoproteins is often challenging because of several factors such as relatively poor ionization of glycopeptide with respect to the peptide, the presence of heterogenous glycan isomers (glycoforms), the lack of a comprehensive database of glycan structures (including microbial and plant derived structures), and the lack of MS signature fragment ions for the complete structure determination. Even though a number of bioinformatics tools are currently available for glycomics and glycopeptide analysis, accurate determination of highly heterogeneous glycan attachment on peptides is still a challenging task (8,13). Thus, the samples are currently split into two separate workflows for the comprehensive characterization of glycan structure on glycoproteins via glycomic and glycoproteomic analysis. Glycoproteins are first proteolytically cleaved to obtain peptides and glycopeptides. The protease digest is directly used for glycoproteomic analysis by injecting to a LC–MS/MS system with or without enrichment. Proteolysis is also performed for glycomic analysis as a preliminary step before the enzymatic release of N-glycans since the glycan release is more efficient from glycopeptides than from intact or denatured glycoproteins because of the decreased steric hindrance (14,15). Nonhuman modifications, such as Galα1-3Gal and N-glycolylneuraminic acid (NeuGc), that could arise from mammalian expression systems can also be determined by both glycomics and glycoproteomics analysis.
Glycomics Analysis
Glycomics analysis enables the introduction of analytes directly into the MS instrument, so multiple MS fragmentation of the analyte ions is possible. Moreover, glycomics allows derivatization of molecules with chromophores, fluorophores, and permethylation, making them more suitable for further downstream analysis techniques such as high performance liquid chromatography (HPLC), NMR, and MS (Figure 2) (16). For the glycomics analysis of glycoproteins, glycans are released by either enzymatic or chemical treatment, depending on the type of glycans being released. N-Glycans are usually released from the glycopeptides using N-glycanase enzymes-for example, either PNGase F or PNGase A, which cleaves the N-linked glycans from the peptide asparagines (17).
The hydrophilic released N-glycans are separated from the peptides and O-linked glycopeptides using a C18 solid-phase extraction (SPE) cartridge or nonporous graphitized carbon column (18). The separated N-glycans are further derivatized based on the downstream analytical technique used for their characterization. The releasing of an O-glycan is usually conducted using chemical methods such as reductive β-elimination, ammonia-based nonreductive β-elimination, or hydrazinolysis since deglycosylation enzymes with wide specificity for O-linked glycans are not available (14,19,20). Similar to N-glycans, released O-linked glycans are also derivatized before downstream analysis, either by permethylation or by reducing-end labeling with chromophores such as 2-aminobenzamide (2-AB), 2-aminopyridine (2-AP), 4-aminobenzoic acid, or anthranilic acid (21).
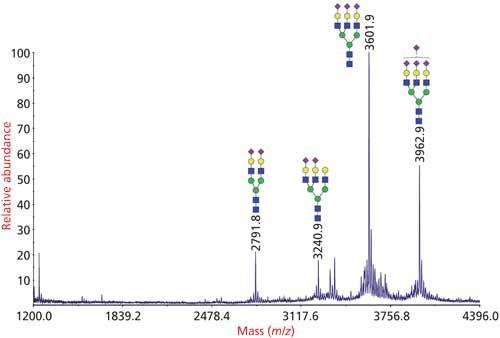
Derivatization enhances the ionization efficiency of the released glycans, and permethylation is the most popular mode of glycan derivatization because it enables detailed structural information of glycans by MSn through both glycosidic and cross-ring cleavages (Figure 3) (22). Moreover, the permethylated glycans can also be further manipulated and used for the determination of glycosylation linkages by GC–MS. For the linkage determination, the permethylated glycans are acid-hydrolyzed, reduced, acetylated, and the resulting PMAAs are analyzed by GC–MS (7).


Figure 3: Analysis of N-glycans from bovine fetuin by glycomics analysis. N-glycans were released from the glycoprotein by PNGase F, permethylated and analyzed by MALDI-TOF-MS.
To quantitate the monosaccharide composition of glycans, monosaccharides derived from glycans by acidic methanolysis were derivatized by trimethylsilyl (TMS) groups and analyzed by GC–MS (23). The monosaccharide composition analysis is also determined by high-performance anion exchange chromatography with pulsed amperometric detection (HPAEC-PAD) of monosaccharides released from the glycans by acid hydrolysis (24).
One of the major drawbacks of the glycomics approach is that the site-specific information of glycosylation, such as the attachment site and occupancy rates, is lost since the glycans are released from the protein. Attempts to perform detailed structural characterization of glycans while keeping the glycan point of attachment to the protein intact are gaining a lot of attention from researchers recently because that analysis simplifies the current multistep analytical procedure used for the characterization of glycosylations.
Glycoproteomics Analysis
The glycans are not released in the glycoproteomics approach and the glycan-peptide bonds are kept intact to obtain information about glycosylation sites and site occupancies. The analysis of intact glycopeptides by LC–MS/MS is the most popular method for the rapid determination of glycosylation at specific site of peptides (Figure 4). Glycoproteomic analysis consists of glycosylation site mapping and determination of the composition of glycans attached at each site (Figures 2, 4, 5, and 6) (17).


Figure 4: Glycoforms of N-linked glycosylated peptide determined by glycoproteomics analysis where the protease digest of the glycoprotein is injected to an LC–MS/MS system and analyzed via HCD–CID fragmentation.
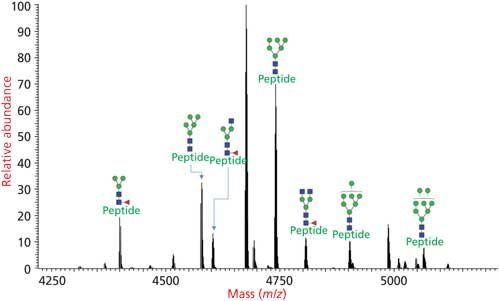
The glycoproteins are digested into smaller peptides and glycopeptides using proteases and the resulting protease digest is injected directly to an LC–MS/MS system. The peptides and glycopeptides fractionated by the LC system is injected to a high-resolution MS instrument and their precursor mass along with the mass of ions after MS fragmentations are analyzed.
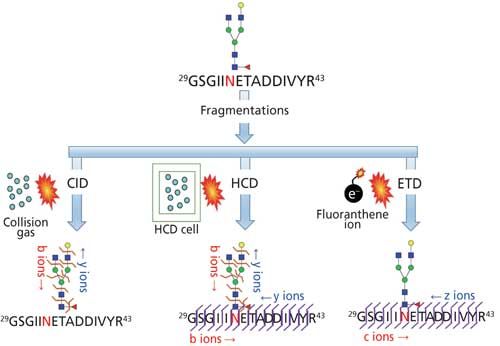
Site mapping reveals the potential glycosylation sites that are occupied and this information is useful for subsequent glycopeptide analysis. Analytical challenges associated with determining the glycosylation site from intact glycopeptides is the lack of adequate peptide fragmentation during MS/MS, thus performing analysis on deglycosylated peptides or partially deglycosylated peptides is required. One of the common techniques for deglycosylation is the enzymatic removal of N-linked glycans with peptide-N-glycosidase (PNGase) in 18O-labeled water (Figure 5a) or partial enzymatic degradation of the N-linked structures using endo-β-N-acetylglucosaminidase (17,25). However, the sites of O-linked glycans can be determined without releasing them since O-glycans are usually smaller in size; peptide fragmentation by a non-ergodic fragmentation approach such as electron transfer dissociation (ETD) can be used for the site determination (Figure 6). Ergodic fragmentation techniques, such as collision-induced dissociation (CID) fragmentation or high energy collisional fragmentation (HCD), will lead to fragmentation of the peptide–glycan bond preferentially, so they are not ideal for the site mapping of O-linked glycopeptides (Figure 7). Moreover, for the site mapping of glycoproteins carrying large or heavily glycosylated (that is, mucin), the removal of O-linked glycans may be required (17). An approach to accomplish the release of O-glycans with simultaneous site-labeling termed β-elimination by Michael addition with dithiothreitol (BEMAD) is also used for site mapping in which a mildly alkaline β-elimination in the presence of dithiothreitol (DTT) is performed (Figure 5b) (26).


Figure 5: Site determination of (a) N-linked glycosylation through 18O labeling by deglycosylation using PNGase F in H218O; (b) O-linked glycosylation through DTT labeling via BEMAD reaction (TEA: triethylamine, DTT: dithiothreitol).
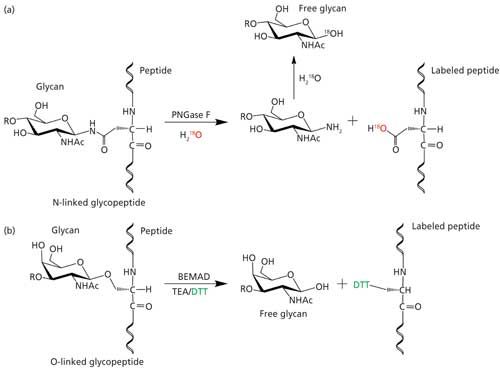


Figure 6: Site mapping of O-glycosylation by ETD MS2 of glycopeptide. The exact location of glycosylation can be mapped from the c and z ion fragments of the ETD spectrum.
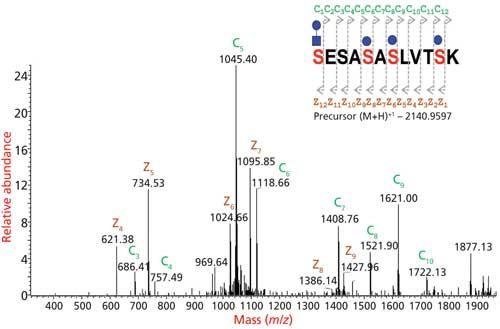


Figure 7: Glycopeptides are analyzed by tandem MS fragmentations. CID provides information on the glycan portion, HCD provides information on glycan portion and partial information on peptide sequence, and ETD provides the site of glycosylation information.
Glycopeptides or deglycosylated and labeled peptides are analyzed by MS (that is, MALDI-TOF MS or ESI-MS) directly or through LC–MS/MS. For the LC–MS/MS analysis, the peptides, glycopeptides, or labeled peptides are first separated on a LC and then injected online into a high-resolution MS system for the mass analysis of intact and fragmented peptide–glycopeptide ions. Information about both the glycans and their attachment sites is obtained from the glycoproteomics data. However, comprehensive characterization of glycans attached on each site is accomplished by the glycomics.
The information about the type of glycans through the glycomic analysis and the site mapping data helps for the accurate and easy glycoproteomic data analysis by narrowing the range of possible masses to look for from the LC–MS/MS data (22). One of the most crucial steps in both glycomics and glycoproteomics analysis is the data interpretation from multiple types of tandem MSn and LC–MS/MS data. Various bioinformatics tools comprising several databases curated through experimental data, an in-silico fragmentation prediction tool, search algorithms, annotation tools, and glycan structure drawing tools are used for the determination of glycosylation on glycoproteins (27–29).
Recent Advances in Glycomics and Glycoproteomics
The complexity and microheterogeneity of glycosylation and the discovery of novel glycosylation from prokaryotes demands methodological progress in the techniques used for the structural characterization of glycoproteins.
Considerable advances have been made recently for the MS analysis of glycoproteins including novel sample preparation techniques such as fractionation, preconcentration; quantitation techniques, such as the use of label-free and isotopic-labeling methods; and instrumentation methods such as ion mobility mass spectrometry (IM-MS) and the flexible use of fragmentation modes. Researchers are increasingly focusing on the quantitative analysis of glycoproteins, in addition to the qualitative data (17).
Better and robust software and computer aided tools were developed recently for the data analysis of highly complex and enormous data volumes obtained through these modern techniques. Volumetric and sampling errors were reduced, and overall reproducibility and analytical throughput were improved by automating the individual steps in glycomics and glycoproteomics. Nevertheless, because of the highly heterogenous nature and presence of very low levels in comparison to cellular proteomes, a comparison of “normal” versus “aberrant” glycosylation levels of complex glycans from various sources is still challenging (30). Glycoprotein characterization using a 96-well plate format via multistep procedures including protein denaturation, deglycosylation, desialylation, permethylation, and subsequent MALDI-MS profiling, were recently achieved successfully (31). Moreover, glycan derivatizations before MS analysis were also automated recently, with very good reproducibility (32). Recently, the permethylation protocol was automated and high-throughput analysis for the glycan profiling of monoclonal antibodies and recombinant human erythropoietin was conducted using robotics (33).
Currently, wide-ranging databases of anti-glycan reagents, such as lectins and antibodies, are available and commercial availability of these reagents for glycoprotein fractionation and glycan-epitope detection have been increased progressively (34–36).
Several recent studies addressed the shortcomings of chemical and enzymatic release of glycans by the development of new chemical glycan release methods (37–39), immobilization of PNGase F (40), optimization of PNGase F release of N-glycans (41), discovery of broad substrate-specific N-glycosidases (42), and high-throughput glycan releasing (30).
Hydrophilic-interaction chromatography (HILIC) HPLC hyphenated with fluorescence detection of reductively aminated glycans with fluorescent tags is the most widely used technique for glycan quantification in the pharmaceutical industry, and the procedure can be validated easily under good manufacturing practice (GMP) regulations. Various recent advances were reported on the development of better HILIC-based separation techniques for the improved isomeric separation of glycans (43–45). Derivatization of glycans with a fluorophore has several advantages, such as enhanced sensitivity of analysis with both spectroscopic and MS detectors and increased hydrophobicity of glycans, thereby increasing their chromatographic retention in reversed-phase LC. A newly reported label, RapiFluorMS (RFMS), enables rapid labeling of released N-glycans at their reducing end. The label bears a quinone moiety as fluorophore and a tertiary amine for strong positive-mode ionization (46).
Much attention has recently shifted in the field of glycomics and glycoproteomics toward quantitative estimation in which MS-based relative and absolute quantification of glycoconjugates is performed. Labeling the analyte with an isotope tag is the most common method for MS-based relative quantitation since the isotope tag does not interfere with chromatography and ionization in MS and provides an isotopic mass shift to distinguish the labeled molecules (47). For the estimation of relative quantity, “light” and “heavy” isotope-labeled glycans, in which isotopic tags with lower mass and higher mass are used, respectively, are mixed at different ratios, and the corresponding MS peak intensity is compared (48).
Even though MALDI and ESI are the most common modes of ionization in glycan and glycopeptide characterization, each of them have their own disadvantages. ESI-MS has the disadvantage of in-source fragmentation, which leads to misinterpretation and poor sensitivity. Several new technologies were developed recently to address these limitations; notably, subambient pressure ionization with a nano-electrospray (SPIN) source was developed in which the ESI emitter was moved to the first vacuum stage of the mass spectrometer at the entrance of the electrodynamic ion funnel to enhance the collection of the entire electrospray plume (49). To optimize the collision energy required for the fragmentation, stepping of CID collision energy that allows simultaneous acquisition of MS/MS spectra of glycopeptide at lower and higher collision energies was developed (50).
Since the glycopeptide MS data interpretation is typically challenging and the false discovery rates (FDR) need to be reduced, intelligent data-dependent decision trees of sequential fragmentation steps of glycopeptides like HCD-product-dependent ETD and CID workflows using orbital ion trap mass spectrometers were developed recently. Such improved MS/MS data and several newly developed data analyzing programs and search engines facilitated data interpretation (30,51).
Other recent developments in the charaterization of glycoproteins include the following advances: improved spectral data of glycans provided by the introduction of novel fragmentation methods such as ultraviolet photo-dissociation (UVPD) (52), the development of IM-MS and the application of it in the discrimination of linkage and position isomers, glycosylation site identification, the identification of α-2-3 and α-2-6 linked sialic acid linkage isomers (53), and the use of capillary zone electrophoresis for the efficient separation, resolution, and sensitivities in the analysis of glycoconjugates (32). Furthermore, an initiative termed minimum information required for a glycomic experiment (MIRAGE) was established in 2011 to promote critical evaluation of experimental protocols, dissemination of data sets for reproducibility, and comparison of results obtained in different laboratories (54).
Summary
Considerable advances were observed during the past decade for the analysis of glycoproteins. The demand for the identification and characterization of the glycome associated with proteins is increasing since the role that protein glycosylation plays in cellular physiology and disease processes is being increasingly deduced. The discovery of novel disease biomarkers, characterization of recombinant glycoprotein therapeutics, the study of the roles of glycosylation on cell signaling and immunology, and microbial and plant glycobiology are the most important fields in which the structural characterization of glycoconjugates is required.
This article has emphasized the most common techniques involved in the interpretation of glycan structure on glycoproteins and also highlighted the recent progress in the field of glycoprotein analysis by mass spectrometry. Advancements in the analytical procedures in glycomics and glycoproteomics would enable rapid yet comprehensive characterization of highly heterogenous glycomes.
Acknowledgments
This work was supported in part by National Institutes of Health (NIH)-funded Research Resource for Biomedical Glycomics (S10OD018530, P41GM103490, R21GM122633) and the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, U.S. Department of Energy grant (DE-SC0015662) at the Complex Carbohydrate Research Center.
Conflict of Interest Statement
The authors work at the Analytical Services & Training Laboratory at the Complex Carbohydrate Research Center at the University of Georgia and perform glycomics and glycoproteomics analysis as collaboration or fee-for-service.
References
- R. Apweiler, H. Hermjakob, and N. Sharon, Biochim. Biophys. Acta 1473, 4–8 (1999).
- R.D. Astronomo and D.R. Burton, Nat. Rev. Drug Discov.9, 308–324 (2010).
- R. Raman, S. Raguram, G. Venkataraman, J.C. Paulson, and R. Sasisekharan, Nat. Methods2, 817–824 (2005).
- M. Guerrini, R. Raman, G. Venkataraman, G. Torri, R. Sasisekharan, and B. Casu, Glycobiology12, 713–719 (2002).
- G. Venkataraman, Z. Shriver, R. Raman, and R. Sasisekharan, Science 286, 537–542 (1999).
- A. Varki, R.D. Cummings, J.D. Esko, P. Stanley, G. Hart, M. Aebi, A. Darvill, T. Kinoshita, N.H. Packer, J.J. Prestegard, R.L. Schnaar, and P.H. Seeberger, in Essentials of Glycobiology, 3rd Edition (Cold Spring Harbor, New York, 2015).
- W. Morelle and J.C. Michalski, Nat. Protoc.2, 1585–1602 (2007).
- M. Wuhrer, M.I. Catalina, A.M. Deelder, and C.H. Hokke, J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 849, 115–128 (2007).
- D.J. Harvey, Mass Spectrom. Rev.18, 349–450 (1999).
- C.E. Costello, J.M. Contado-Miller, and J.F. Cipollo, J. Am. Soc. Mass Spectrom.18, 1799–1812 (2007).
- Y. Pu, M.E. Ridgeway, R.S. Glaskin, M.A. Park, C.E. Costello, and C. Lin, Anal. Chem. 88, 3440–3443 (2016).
- H. Ito, H. Kaji, A. Togayachi, P. Azadi, M. Ishihara, R. Geyer, C. Galuska, H. Geyer, K. Kakehi, M. Kinoshita, N.G. Karlsson, C. Jin, K. Kato, H. Yagi, S. Kondo, N. Kawasaki, N. Hashii, D. Kolarich, K. Stavenhagen, N.H. Packer, M. Thaysen-Andersen, M. Nakano, N. Taniguchi, A. Kurimoto, Y. Wada, M. Tajiri, P. Yang, W. Cao, H. Li, P.M. Rudd, and H. Narimatsu, Glycoconj. J. 33, 405–415 (2016).
- L. Cao, Y. Qu, Z. Zhang, Z. Wang, I. Prytkova, and S. Wu, Expert Rev. Proteomics13, 513–522 (2016).
- W.R. Alley, Jr. and M.V. Novotny, Annu. Rev. Anal. Chem.6, 237–265 (2013).
- Z. Szabo, A. Guttman, and B.L. Karger, Anal. Chem.82, 2588–2593 (2010).
- Y. Wada, A. Dell, S.M. Haslam, B. Tissot, K. Canis, P. Azadi, M. Backstrom, C.E. Costello, G.C. Hansson, Y. Hiki, M. Ishihara, H. Ito, K. Kakehi, N. Karlsson, C.E. Hayes, K. Kato, N. Kawasaki, K.H. Khoo, K. Kobayashi, D. Kolarich, A. Kondo, C. Lebrilla, M. Nakano, H. Narimatsu, J. Novak, M.V. Novotny, E. Ohno, N.H. Packer, E. Palaima, M.B. Renfrow, M. Tajiri, K.A. Thomsson, H. Yagi, S.Y. Yu, and N. Taniguchi, Mol. Cell Proteomics 9, 719–727 (2010).
- A. Shajahan, C. Heiss, M. Ishihara, and P. Azadi, Anal. Bioanal. Chem.409, 4483–4505 (2017).
- Y. Li, J. Fu, Y. Ling, T. Yago, J.M. Mcdaniel, J. Song, X. Bai, Y. Kondo, Y. Qin, C. Hoover, S. Mcgee, B. Shao, Z. Liu, R. Sonon, P. Azadi, J.D. Marth, R.P. Mcever, C. Ruan, and L. Xia, Proc. Natl. Acad. Sci. U. S. A. 114, 8360–8365 (2017).
- I. Turyan, X. Hronowski, Z. Sosic, and Y. Lyubarskaya, Anal. Biochem.446, 28–36 (2014).
- R.P. Kozak, L. Royle, R.A. Gardner, D.L. Fernandes, and M. Wuhrer, Anal. Biochem.423, 119–128 (2012).
- D.J. Harvey, J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 879, 1196–1225 (2011).
- W.R. Alley, Jr., B.F. Mann, and M.V. Novotny, Chem. Rev.113, 2668–2732 (2013).
- J. Bleton, P. Mejanelle, J. Sansoulet, S. Goursaud, and A. Tchapla, J. Chromatogr. A720, 27–49 (1996).
- M.R. Hardy and R.R. Townsend, Methods Enzymol.230, 208–225 (1994).
- B. Kuster and M. Mann, Anal. Chem.71, 1431–1440 (1999).
- L. Wells, K. Vosseller, R.N. Cole, J.M. Cronshaw, M.J. Matunis, and G.W. Hart, Mol. Cell Proteomics1, 791–804 (2002).
- M. Bern, Y.J. Kil, and C. Becker, Curr. Protoc. Bioinformatics Chapter 13, Unit13 20 (2012).
- A. Ceroni, K. Maass, H. Geyer, R. Geyer, A. Dell, and S.M. Haslam, J. Proteome Res.7, 1650–1659 (2008).
- M. Eavenson, K.J. Kochut, J.A. Miller, R. Ranzinger, M. Tiemeyer, K. Aoki, and W.S. York, Glycobiology25, 66–73 (2015).
- S. Gaunitz, G. Nagy, N.L.B. Pohl, and M.V. Novotny, Anal. Chem. 89, 389–413 (2017).
- M.R. Bladergroen, K.R. Reiding, A.L.H. Ederveen, G.C.M. Vreeker, F. Clerc, S. Holst, A. Bondt, M. Wuhrer, and Y.E.M. Van Der Burgt, J. Proteome Res. 14, 4080–4086 (2015).
- L.R. Ruhaak, C.a.M. Koeleman, H.W. Uh, J.C. Stam, D. Van Heemst, A.B. Maier, J.J. Houwing-Duistermaat, P.J. Hensbergen, P.E. Slagboom, A.M. Deelder, and M. Wuhrer, Plos One 8, e73082 (2013).
- A. Shubhakar, R.P. Kozak, K.R. Reiding, L. Royle, D.I. Spencer, D.L. Fernandes, and M. Wuhrer, Anal. Chem.88, 8562–8569 (2016).
- S. Fanayan, M. Hincapie, and W.S. Hancock, Electrophoresis33, 1746–1754 (2012).
- Y.L. Hu, S.Y. Zhou, C.Y. Yu, H.X. Tang, and Y. Mechref, Rapid Commun. Mass Spec.29, 135–142 (2015).
- M.J. Kailemia, L.R. Ruhaak, C.B. Lebrilla, and I.J. Amster, Anal. Chem.86, 196–212 (2014).
- W.X. Chen, J.M. Smeekens, and R.H. Wu, J. Proteome Res.13, 1466–1473 (2014).
- X.Z. Song, H. Ju, Y. Lasanajak, M.R. Kudelka, D.F. Smith, and R.D. Cummings, Nat. Methods 13, 528–534 (2016).
- X.Z. Song, H. Ju, C.M. Zhao, and Y. Lasanajak, Bioconjugate Chem. 25, 1881–1887 (2014).
- M. Szigeti, J. Bondar, D. Gjerde, Z. Keresztessy, A. Szekrenyes, and A. Guttman, J. Chromatogr. B 1032, 139–143 (2016).
- Y.J. Weng, Z.G. Sui, H. Jiang, Y.C. Shan, L.F. Chen, S. Zhang, L.H. Zhang, and Y.K. Zhang, Sci Rep-Uk5, 9770 (2015).
- Y.M. Du, T. Xia, X.Q. Gu, T. Wang, H.Y. Ma, J. Voglmeir, and L. Liu, J. Agr. Food Chem.63, 10550–10555 (2015).
- G.O. Guerrant and C.W. Moss, Anal. Chem.56, 633–638 (1984).
- G.Y. Qing, X.L. Li, P. Xiong, C. Chen, M.M. Zhan, X.M. Liang, and T.L. Sun, ACS Appl. Mater. Inter.8, 22084–22092 (2016).
- W.Y. Shao, J.X. Liu, K.G. Yang, Y. Liang, Y.J. Weng, S.W. Li, Z. Liang, L.H. Zhang, and Y.K. Zhang, Talanta158, 361–367 (2016).
- M.A. Lauber, Y.Q. Yu, D.W. Brousmiche, Z.M. Hua, S.M. Koza, P. Magnelli, E. Guthrie, C.H. Taron, and K.J. Fountain, Anal. Chem.87, 5401–5409 (2015).
- C.M. Woo, A.T. Iavarone, D.R. Spiciarich, K.K. Palaniappan, and C.R. Bertozzi, Nature Methods12, 561–567 (2015).
- J.A. Atwood, L. Cheng, G. Alvarez-Manilla, N.L. Warren, W.S. York, and R. Orlando, J. Proteome Res.7, 367–374 (2008).
- J.T. Cox, S.R. Kronewitter, A.K. Shukla, R.J. Moore, R.D. Smith, and K.Q. Tang, Anal. Chem.86, 9504–9511 (2014).
- H. Hinneburg, K. Stavenhagen, U. Schweiger-Hufnagel, S. Pengelley, W. Jabs, P.H. Seeberger, D.V. Silva, M. Wuhrer, and D. Kolarich, J. Am. Soc. Mass Spectr. 27, 507–519 (2016).
- S.W. Wu, T.H. Pu, R. Viner, and K.H. Khoo, Anal. Chem. 86, 5478–5486 (2014).
- B.J. Ko and J.S. Brodbelt, Int. J. Mass Spectrom.377, 385–392 (2015).
- C.J. Gray, B. Thomas, R. Upton, L.G. Migas, C.E. Eyers, P.E. Barran, and S.L. Flitsch, BBA-Gen Subjects1860, 1688–1709 (2016).
- D. Kolarich, E. Rapp, W.B. Struwe, S.M. Haslam, J. Zaia, R. Mcbride, S. Agravat, M.P. Campbell, M. Kato, R. Ranzinger, C. Kettner, and W.S. York, Mol. Cell. Proteomics12, 991–995 (2013).
Asif Shajahan and Parastoo Azadi are with the Complex Carbohydrate Research Center at the University of Georgia in Athens, Georgia. Direct correspondence to: azadi@ccrc.uga.edu















Mass Spectrometry for Forensic Analysis: An Interview with Glen Jackson
November 27th 2024As part of “The Future of Forensic Analysis” content series, Spectroscopy sat down with Glen P. Jackson of West Virginia University to talk about the historical development of mass spectrometry in forensic analysis.

Detecting Cancer Biomarkers in Canines: An Interview with Landulfo Silveira Jr.
November 5th 2024Spectroscopy sat down with Landulfo Silveira Jr. of Universidade Anhembi Morumbi-UAM and Center for Innovation, Technology and Education-CITÉ (São Paulo, Brazil) to talk about his team’s latest research using Raman spectroscopy to detect biomarkers of cancer in canine sera.



