Mid-IR Spectroscopy as a Primary Tool in Carbon Biogeochemistry Research
Special Issues
Mid-infrared spectroscopy is applied as a primary technique to better understand the bio- and ultraviolet-transformation of soil microbial biomass. Soil microbial biomass constitutes as much as 50% organic carbon in soil organic matter, and therefore plays a crucial role in soil-atmospheric chemistry. In this study, the spatial distribution of microbial-derived organic structures on kaolinite and montmorillonite clay minerals was investigated.
Previous studies have shown that soil microbial biomass constitutes as much as 50% organic carbon in soil organic matter, and therefore plays a crucial role in soil-atmospheric chemistry and climate. However, little is known about the fate and transformation of the magnitude of microbial components in the soil. In this study, mid-infrared (IR) spectroscopy is applied as a primary technique to better understand the bio- and ultraviolet-transformation of soil microbial biomass. The spatial distribution of microbial-derived organic structures on kaolinite and montmorillonite clay minerals was investigated to decipher which organic structures preferentially associate with the crystalline and amorphous forms of the minerals, which may be accessible to microbial heterotrophs, and which are physically protected from decomposition.
Soil organic matter (SOM) is a complex mixture of compounds with varied sources, compositions, and environmental and ecological roles. Generally, the biogeochemical characteristics of SOM represent an integrated signal of multiple processes, including degradation and its interaction with soil minerals and pedogenic oxides. Recently, the microbial contribution to SOM has been shown to be much larger than previously thought, and therefore its role in the carbon and nitrogen biogeochemical cycles may also be underestimated (1,2). Considering the huge amounts of carbon stored in soils, and the unprecedented pressure on land due to rapid population growth, climate change, land degradation, and other environmental stressors, there is an urgent need to better understand the degradation dynamics of SOM, as well as its interactions with pristine and structurally altered (weathered) clay minerals.
In soils, biological recalcitrance of organic matter (OM) can be attained through molecular structure and formation of cross linkages, but organic matter–mineral interactions have also been shown to play a crucial role in the preservation and distribution of SOM. Overall, organic matter–mineral interactions have significant implications for a number of biogeochemical processes, ranging from the formation, growth, and division of the earliest cells, to agricultural productivity and other ecosystem services, such as enhancing the ability of soils to act as a natural modulator of global climate change (3,4). Despite these important roles and functions, molecular-level information about the mechanisms of such interactions remains poorly understood, and even less is known about the effects of micro-scale weathering of minerals on these interactions. Therefore, these gaps in our knowledge may serve to undermine our ability to predict future carbon cycling under a warming climate, and to reconstruct paleo-environmental conditions based on organic matter-derived proxies.
In this contribution, mid-infrared (IR) spectroscopy is applied as a primary technique to better understand the bio- and ultraviolet-transformation of soil microbial biomass. Further, the spatial distribution of microbial-derived organic structures on kaolinite (1:1) and montmorillonite (2:1) clay minerals is investigated, in order to decipher which organic structures preferentially associate with the crystalline and amorphous (weathered) forms of the minerals, which may be accessible to microbial heterotrophs, and which are physically protected from decomposition. IR spectroscopy provides a rapid nondestructive means to perform molecular-level investigations on SOM and their interactions with soil minerals and pedogenic oxides. Critically, since samples are not subjected to chemical treatment, there are no side effects of secondary reactions. Additionally, the molecular composition of the sample is determined simultaneously, which simplifies and shortens the time required for spectral processing, including the quantitative determination of major macromolecular components (5).
Materials and Methods
Microbial Propagations and Conditions
A mixed heterotrophic soil biome was prepared in trypicase soy broth (TSB), supplemented with 0.5% (w/v) yeast extract. Initially, 1 g of freshly collected, homogenized agricultural soil was added to 100 mL aliquots of sterile distilled water, and dispersed by stirring. Aliquots (1 mL) of the initial soil suspension were used to prepare serial dilutions of 10-4 g soil suspension in a mixture of TSB–yeast extract and sterile distilled water (1:9). Finally, 1-mL aliquots from the serial dilutions were used to inoculate 100 mL of the undiluted growth solution, and the cultures incubated at room temperature for 72 h, with shaking at 150 rpm. Dilutions were done to prevent any possible carryover of soil particles from the initial soil suspension. After incubation, the cultures were harvested by centrifugation, and the cell pellets washed in ultrapure water and freeze-dried (6,7).
Microbial Degradation
Bio- and ultraviolet degradation of soil microbial biomass and biodegradation of kaolinite-microbial complex were conducted as described by Spence and Kelleher (8). Biodegradation was carried out under ambient conditions for 26 weeks.
Microscale Weathering of Clay Material
Microscale weathering of untreated (raw) montmorillonite (SWy-2; Source Clays Repository; The Clay Minerals Society) was carried out for 36 h, according to the protocol described by Spence and associates (9).
Adsorption Experiment
Duplicate clay-microbial adsorption experiments were carried using raw kaolinite, as well as raw and acid-treated (weathered/degraded) montmorillonite clay minerals (9).
IR Analysis
The IR spectra of initial and degraded (bio- and ultraviolet) biomass, raw kaolinite, raw and acid-treated montmorillonite, and their related microbial complexes were acquired on a Bruker Tensor Series FT-IR spectrometer, using the KBr pressed disk technique. Thoroughly dried samples (~1 mg) were homogenized in 100 mg of spectroscopic-grade KBr (Sigma). Background KBr spectra were obtained and spectra ratioed to the background. Spectra were recorded by accumulating 256 scans in the 4000 to 400 cm-1 mid-IR spectral range in the absorbance mode, with a resolution of 4 cm–1. To minimize complications arising from unavoidable spectral shifts, baseline correction was applied to all spectra using the automatic baseline correction method. In addition to the use of thoroughly dried samples, it was essential to analyze the disk-like pellets immediately after preparation. This was done to minimize the adsorption of atmospheric water by the hygroscopic KBr, hence reducing the possibility of chemical exchange between the clay and KBr, which could result in a change of the crystalline structure of the mineral (10). Further, rapid analysis of the pellets was important to eliminate the suppression of key signals from the samples by KBr-adsorbed water.


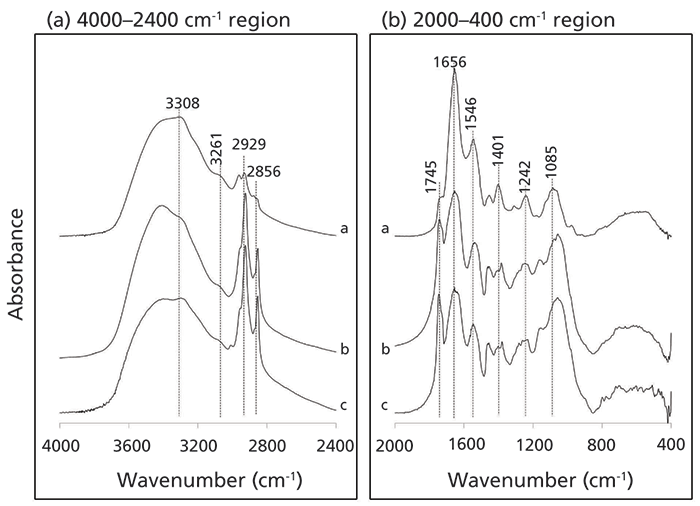
Figure 1: Stacked infrared spectra of (a) fresh microbial biomass; (b) microbial biomass degraded under ambient light for 26 weeks; and (c) microbial biomass degraded under ultraviolet light for 26 weeks. The spectra are presented to highlight the 4000–2400 cm-1 and the 2000–400 cm-1 spectral regions. The spectra in each box are offset and shown with similar scaling so that clear comparisons can be made.
Results and Discussion
Figure 1 presents the IR spectra of fresh microbial biomass (a), and that degraded under ambient (b) and ultraviolet (c) conditions. General peak assignments are applicable to all spectra, and the characteristic functionalities of major biochemical components are summarized in Table I. After degradation has occurred, major structural changes are observed, particularly the enrichment of polymethylenic-C [(CH2)n] in long-chained aliphatic structures (CHx stretching bands at 3000 cm–1 to 2700 cm-1), indicating that they are selectively preserved as part of the stable carbon pool. Signals from other biochemical components (proteins and carbohydrates; see Table I) are attenuated, indicating that they have degraded relative to aliphatics. Overall, there is no distinction between bio- and photodegradation products. Bio- and photodegradation are primary decomposition processes involved in organic matter transformation in the terrestrial environment (8).


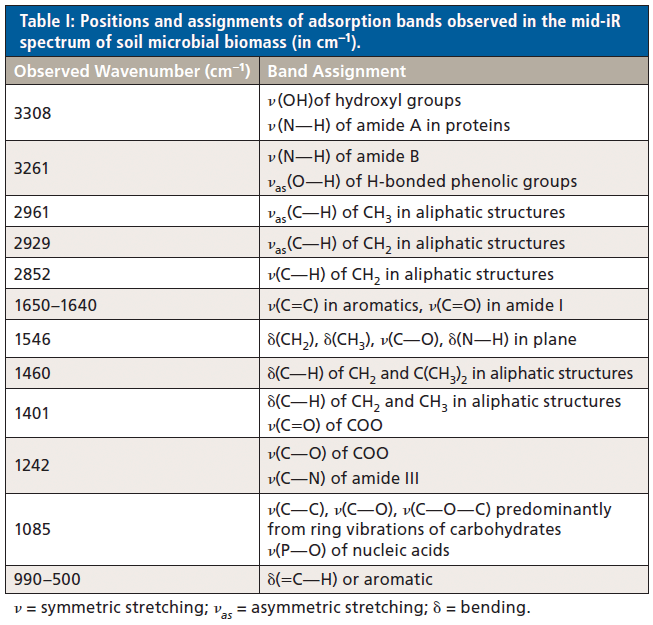
The superimposed spectra of raw and acid-treated montmorillonite are presented in Figure 2. A band at 3626 cm-1 denotes v(OH) of structural OH groups coordinated to octahedral cations (Al–O[OH]); Al-Al-OH bending (917 cm–1), Al-Fe-OH (881 cm–1), and Al-Mg-OH (844 cm–1), suggesting that octahedral Al3+ is partially substituted by Fe2+ or Mg2+ ions (11). Two shoulders near 881 cm–1 and 844 cm–1 suggest a relatively low concentrations of Fe2+ and Mg2+ ions, respectively. After acid-treatment, the v(OH) band (3626 cm-1), the –OH bending vibrations near 917, 881, and 844 cm–1, and δ(Si–O–Al) at 526 cm–1 (12) are no longer observed, demonstrating a depletion in the content of Al3+ and substituted Mg2+ and Fe2+ cations in the octahedral sheet. This is supported by the attenuation of spectral hydration peaks in the –OH stretching (3419 cm–1) and v2(H2O) bending regions 1650–1623 cm–1, and an incremental shift in the position of the v2(H2O) peak from 1623 to 1620 cm–1. A shoulder near 3225 cm–1, indicating an overtone (2v2) of bending mode of cation hydration water in the raw mineral, is not observed in the acid-treated clay. Microscale weathering of the tetrahedral (Si environment) is also observed, and is characterized by changes in both the shape and position of the Si–O stretching band (1047 cm–1), now appearing at 1095 cm–1, and the enrichment of the asymmetric band 790 cm–1. This would suggest that acid hydrolysis (a key process in soil formation) has transformed the crystalline structure of montmorillonite to amorphous silica. All remaining bands (1095, 790, and 463 cm–1 [12]) in the acid-treated spectrum are synonymous with Si–O vibrations.


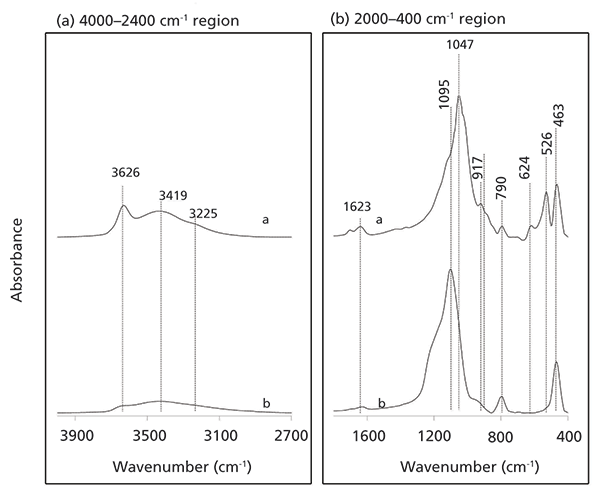
Figure 2: Infrared spectra of (a) raw montmorillonite, and (b) montmorillonite samples treated with 6 mol/L of HCl for 36 h. The spectra are presented to highlight the 4000–2400 cm-1 and the 2000–400 cm-1 spectral regions.
Figure 3 illustrates the interactions of microbial biomass with raw and weathered montmorillonite. Microbial interactions with raw montmorillonite is dominated by signals from aliphatic and proteinaceous components (Figure 3a; see Table I for full assignment of spectral peaks). The results also clearly demonstrate that only limited quantities of microbial-derived components (primarily proteins [1640 cm–1]) are adsorbed to the weathered structure. This would suggest that octahedral and interlayer cations present in a crystalline structure play important roles in the adsorption process.


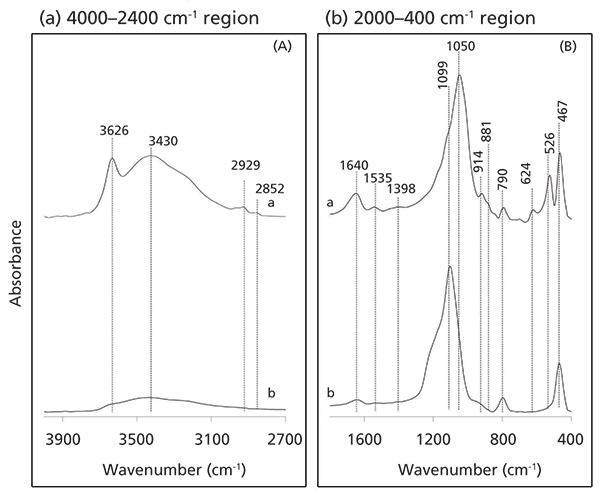
Figure 3: Inset spectra: superimposed infrared spectra of the interactions of a mixed heterotrophic soil biome with (a) raw montmorillonite, and (b) sample treated with 6 mol/L HCl for 36 h. The spectra are presented to highlight the 4000–2400 cm-1 and the 2000–400 cm-1 spectral regions.
The IR spectra of raw kaolinite (a), kaolinite–microbial complex (b), and the latter sample degraded under ambient conditions for 26 weeks (c) are presented in Figure 4. Raw kaolinite exhibits four characteristic peaks assigned to v(OH) stretching vibrations of surface OH groups (3652, 3671, and 3694 cm-1), and v(OH) vibrations of inner OH groups (3620 cm-1). The v(Si–O) is represented by three strong, well-resolved bands in the 1100–1000 cm-1 region. Bands at 936 and 913 cm-1 represent OH-bending vibrations of the outer and inner surface OH groups of the mineral, respectively. Additional bands near 701 and 755 cm-1 are associated with the surface OH groups. Bands due to δ(Al-Fe-OH) at 865–875 cm-1 and stretching at 3607 cm-1 are typical of Fe-bearing kaolinites, while the absorption bands due to Si–O–Al and Si–O–Si are observed at 544 and 473 cm-1, respectively (13). After interactions have occurred, the v(OH), δ(OH), v(SiO), and the δ(SiO) bands are now attenuated (Figure 4b and c). Further, the v(OH) band at 3671 cm-1 is no longer observed, suggesting that hydrogen bonding with microbial-derived components has occurred.
Conversely, the bands at 3621, 3652, and 3694 cm-1 remained unchanged, indicating there was no intercalation of the mineral. Signals form v(OH) in phenolic and carboxylic groups and v(N-H) of amides from major biochemical groups appear as a strong broad at 3408–3300 cm-1. Microbial–mineral interactions are further evidenced by peaks at 2929 and 2856 cm-1 assigned to vas(CH2) and v(CH2) from CH2 methylene groups, respectively. When coupled with a weak shoulder at 2960 cm-1 (vas[C–H] of methyl groups), this would suggest a contribution from polymethylenic-C ([C-H2]n]) with a low degree of branching in the interaction process (14). Contributions from other biochemical components with roles in the interaction process, including bands at 1657 cm-1 attributed to aromatic v(C=C) bonds, v(O–C=O) of metal-coordinate carboxylates, H-bonded v(C=O) of amides, and amide I band in peptides and proteins. Absorption peaks of moderate to low intensity at 1545, 1425, 1394–1380, and 1235 cm-1 denotes δ(N–H) of amide II of proteins; δ(O-H) of carboxylic acids, the CO2 stretch of carboxylates and the aliphatic CH2 group of alkanes; δ(CH2) and δ(CH3), phenolic v(C-O) or vsym(OCO) of coordinated carboxylate groups; and v(C-O) of carboxylic acid and the v(C-N) of amides (amide III), respectively (14). A weak shoulder at 1099–1095 cm-1 corresponds to the C–O–O and C–O ring vibration of carbohydrates, most likely from bacterial cell wall components such as peptidoglycan. Note, these figures remain largely unchanged following degradation of the clay-microbial complex, suggesting that microbial-derived biomolecules are physically protected.



Figure 4: FT-IR absorbance spectra of (a) pure kaolinite, (b) initial kaolinite-microbial complex, and (c) the latter sample degraded under ambient conditions (4000–400 cm-1 spectral region).
Conclusions
In this study, mid-infrared (IR) spectroscopy is applied as a primary technique to better understand the bio- and ultraviolet-transformation of soil microbial biomass. The spatial distribution of microbial-derived organic structures on kaolinite and montmorillonite clay minerals was investigated to decipher which organic structures preferentially associate with the crystalline and amorphous forms of the minerals, which may be accessible to microbial heterotrophs, and which are physically protected from decomposition.
Bio- and photodegradation produced indistinguishable results, and are both characterized by considerable enrichment in aliphatic components, presumably polymethylenic-C ([C-H2]n), and a concomitant decrease in the relative concentrations of carbohydrate and protein structures in a clay-free environment. Results also indicate that microbial-derived organic matter (OM), primarily aliphatic components, adsorbed mainly to the external surfaces of the minerals, but also to the interlaminar region of montmorillonite. Similarly, aliphatic structures appear to be dominant after degradation of OM-mineral complexes had occurred. After chemical weathering of montmorillonite, the adsorption of microbial-derived components (in particular lipids) to the amorphous form of the mineral appears to have decreased. Based on this behavior, an immediate conclusion is that spatial co-variation of microbial-derive OM with octahedral cations in the minerals may be a primary mechanism of interaction.
References
(1) R. Kindler, A. Miltner, H-H. Richnow, and M. Kastner, Soil Biol. Biochem. 38, 2860–2870 (2006).
(2) A.J. Simpson, M.J. Simpson, E. Smith, and B.P. Kelleher, Environ. Sci. Technol. 41, 8070–8076 (2007).
(3) M.S. Huang, Soil Mineral-Organic Matter-Microorganism Interactions and Ecosystem Health: Dynamic, Mobility and Transformation of Pollutants and Nutrients, A. Violante, J-M. Bollag, L. Gianfreda, and P.M. Huang, Eds. (Elsevier Science, Amsterdam, The Netherlands, 2002), pp. 1–36.
(4) P.M. Torn, Biophysico-Chemical Processes Involving Natural Nonliving Organic Matter in Environmental Systems, N. Senesi, B. Xing, and P.M. Huang, Eds. (John Wiley & Sons, Hoboken, New Jersey, 2009), pp. 219–272.
(5) M. Grube, J. Zagreba, E. Gromozova, and M. Fomina, Vib. Spectrosc. 19, 301–306 (1999).
(6) D. Borrok, J.B. Fein, and C.F. Kulpa, Cosmochim. Acta 68, 3231–3238 (2004).
(7) I.V.N Rathnayake, M. Megharaj, G.S.R. Krishnamurti, N. Bolan, and R. Naidu, Chemosphere 90, 1195–1200 (2013).
(8) A. Spence and B.P. Kelleher, J. Mol. Struct. 1107, 7–13 (2016).
(9) A. Spence, C. Robinson, and R.E. Hanson, Mol. Struct. 1056–1057, 157–165 (2014).
(10) N. van Breemen and P. Buurman, Soil Formation (Kluwer Academic Publishers, Dordrecht, The Netherlands, 2000).
(11) J. Madejová and P. Komadel, Clays Clay Miner. 49, 410–432 (2001).
(12) M. Pentrák, V. Bizovská, and J. Madejová, Vib. Spectrosc. 63, 360–366 (2012).
(13) K. Tazaki, ClaysClay Miner. 53, 224–233 (2005).
(14) C.P. Marshall, E.A. Carter, S. Leuko, and E. Javaux, Vib. Spectrosc. 41, 182–189 (2006).
Adrian Spence is with the International Centre for Environmental and Nuclear Sciences, at the University of the West Indies, in Mona, Kingston, Jamaica. Direct correspondence to: adrian.spence02@uwimona.edu.jm













LIBS Illuminates the Hidden Health Risks of Indoor Welding and Soldering
April 23rd 2025A new dual-spectroscopy approach reveals real-time pollution threats in indoor workspaces. Chinese researchers have pioneered the use of laser-induced breakdown spectroscopy (LIBS) and aerosol mass spectrometry to uncover and monitor harmful heavy metal and dust emissions from soldering and welding in real-time. These complementary tools offer a fast, accurate means to evaluate air quality threats in industrial and indoor environments—where people spend most of their time.


NIR Spectroscopy Explored as Sustainable Approach to Detecting Bovine Mastitis
April 23rd 2025A new study published in Applied Food Research demonstrates that near-infrared spectroscopy (NIRS) can effectively detect subclinical bovine mastitis in milk, offering a fast, non-invasive method to guide targeted antibiotic treatment and support sustainable dairy practices.

Smarter Sensors, Cleaner Earth Using AI and IoT for Pollution Monitoring
April 22nd 2025A global research team has detailed how smart sensors, artificial intelligence (AI), machine learning, and Internet of Things (IoT) technologies are transforming the detection and management of environmental pollutants. Their comprehensive review highlights how spectroscopy and sensor networks are now key tools in real-time pollution tracking.


