A Robust and Sensitive Method for Detecting Glyphosate and Other Polar Pesticides in Food and Water: Multiple Analytes in a Single Injection without Derivatization
Special Issues
A new high-throughput LC–MS/MS method meets the challenge of eliminating matrix effects for monitoring, with high specificity, polar organic pesticides such as glyphosate in food and water, while meeting targeted limits of detection.
Monitoring a highly polar, small organic pesticide such as glyphosate in food and water sources presents a significant challenge. Polar pesticides are not amenable to standard extraction procedures, are frequently poor ionizers, and do not separate well. Current analysis methods rely on labor-intensive derivatization and cleanup steps. This study provides details of a robust high-throughput liquid chromatography tandem mass spectrometry (LC–MS/MS) assay for the detection of glyphosate and its metabolites in complex food and water matrices, without the need for derivatization of samples prior to analysis. Polar pesticides are water soluble, and the extraction was therefore aqueous with methanol and formic acid added to improve efficiency. The large variety of interfering particulates in environmental and drinking water samples requires further sample preparation, and a simple filtration step was performed here before the MS/MS analysis. The food analytes were well separated from matrix interferences in most of the foods tested, and all water sample analytes were well retained after removal of interferences. This new method largely eliminated matrix effects and achieved unambiguous identification, reproducible retention times, and sensitivity; and attains the targeted limits of detection.
Despite its association with various health risks, glyphosate remains the most commonly used herbicide worldwide. Glyphosate-based pesticide formulations are used as a simple solution to improve yields by reducing weed growth around glyphosate resistant crops. Almost inevitably, some level of glyphosate residue ends up in the food chain, and it has been detected in the urine of a range of animal species, including humans (1). Polar pesticides are frequently poor ionizers, may suffer from low extraction from the sample matrix, and demonstrate poor chromatographic separation. These pesticides, therefore, have historically required complex single residue methods to make them amenable to analysis, involving time-consuming derivatization steps and considerable clean up procedures.
The analysis of glyphosate using high performance liquid chromatography with fluorescence detection (HPLC–FLD) in water is well known, with or without derivatization. Usually, glyphosate undergoes derivatization by the reaction of the native glyphosate with fluorenylmethyloxycarbonyl chloride (FMOC-Cl) before separation on a reversed-phase column. If the separation is performed on a polar chromatographic column, post-column derivatization before detection is required. The tedious derivatization step complicates the analysis, and the reproducibility is poor. Thus, there is a growing need for a method that can detect not only glyphosate and its major metabolite aminomethylphosphonic acid (AMPA), but also glufosinate and similar highly polar compounds, in their underivatized states.
Modern triple quadrupole mass spectrometers that allow fragmentation analysis offer very low detection limits and high detection selectivity. This approach enables laboratories to rapidly screen samples for a variety of regulated pesticides using the European Union Reference Laboratories' (EURL) quick, easy, cheap, efficient, rugged, and safe (QuEChERS) sample extraction and cleanup method. Owing to this, multi-residue liquid chromatography–tandem mass spectrometry (LC–MS/MS) analyses have become the minimum standard for the quantification of many pesticides in food and water samples. Nevertheless, glyphosate and other highly polar pesticides are not suitable for this type of analysis, and remain challenging for routine screening.
The EURL quick polar pesticides (QuPPe) method allows the simultaneous analysis of a number of highly polar pesticides that are not amenable to common multiresidue methods. However, in practice, the separation is not robust, and requires intense system maintenance. The method involves extraction with acidified methanol and LC–MS/MS measurement, using isotope labeled internal standards for accurate quantification. Ion chromatography is the preferred separation technique for polar ionic analytes, such as anions, cations or small polar analytes (metabolites), and sugars. To use an ion chromatographic approach for the analysis of polar pesticides offers the ability to include multiple analytes in a single injection, without derivatization or the use of an ion suppressor. Food testing laboratories would benefit greatly, in terms of both time and cost, from a methodology that avoids these complex analysis procedures. Here, we present a robust and sensitive method for the direct analysis of polar pesticides in food and environmental samples without derivatization (2).
Experimental
Extraction
Food Samples
The EURL QuPPe method for extraction of polar pesticides from food samples of plant and animal origin is well developed (3). In this experiment, the QuPPe extraction was adjusted by using a little less methanol and more formic acid. Because polar pesticides are water soluble, the extraction was aqueous with methanol and formic acid added to improve efficiency. The addition of internal standards to matrices is considered essential in order to compensate for matrix effects and the shifting retention times observed in many chromatographic separation procedures. Dispersive solid-phase extraction (SPE) cleanup using C18 was also carried out as described in the QuPPe-AO3 method. A push-through method was finalized using two sorbents and SPE filters, because the extracts obtained from the QuPPe method require extensive cleanup for more stable chromatography and less pollution of the source.
The eluents' incompatibility with electrospray ionization sources requires the use of a suppressor, so as to lower the ion load for the electrospray ionization (ESI) source, and gain sensitivity. By employing a polyvinyl alcohol based column with quaternary ammonium groups, and using an ammonium bicarbonate buffer prior to detection by a highly sensitive quadrupole MS system, the need for a suppressor was removed.
Water Samples
Environmental and drinking water samples vary widely in the degree of particle content, causing difficulties for LC injection and the reproducibility. SPE-type cleanup would add significant time, as well as financial cost, in a high throughput laboratory situation where minimal sample preparation is desirable. To overcome these challenges, and to be able to swiftly change between food and environmental samples, the same setup for food samples (except the 500 µL injection volume) was carried out. A simple filtration step, using Chromacol 17-SF-02 (RC) from 17-mm syringe filters, was performed when transferring the samples to the LC vials. Internal standards to a final concentration of 1 ppb were added to samples and standard solutions, and QC samples in tap water were prepared in a similar fashion. To minimize matrix interferences, standard additions to the samples were used.
Separation
Separation was achieved using a Shimadzu Nexera ultrahigh-pressure liquid chromatography (UHPLC) system comprising LC-30AD pumps, a SIL-30AC autosampler fitted with a 500 µL loop, and a CTO-20A column oven. For chromatography, a polyvinyl alcohol column, with quaternary ammonium groups using a bicarbonate eluent between pH 8 and 10, was used.
Food Samples
The finalized chromatographic method used 10 µL injections onto a 150 x 4 mm column, employing a 20 mm guard column of the same material, and a 0.5 µm filter, with a flow rate of 0.6 mL/min. Both columns were replaced every 250 samples to maintain performance, and to keep the MS source clean. Performance was shown to be robust and reproducible on a variety of food matrices used for method verification laid out in Table I, all subject to the cleanup method as described above.


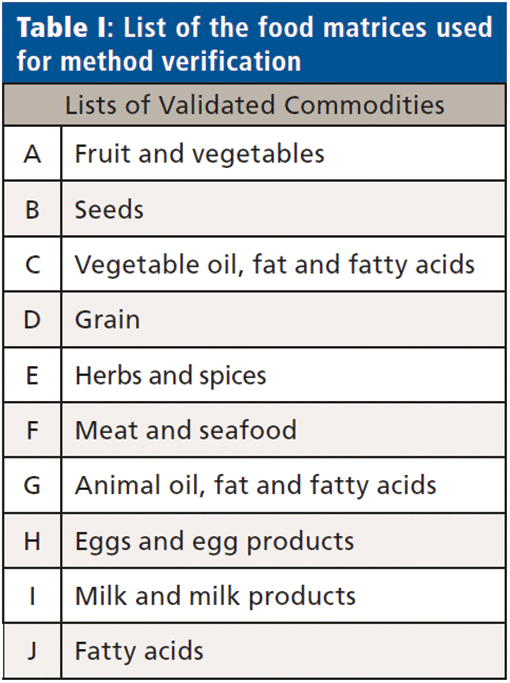
Water Samples
An injection volume of 500 µL was employed in a chromatographic method similar to that used for the food samples.
MS/MS Analysis
Analyses were performed using a triple quadrupole linear ion trap mass spectrometer (Sciex QTRAP 6500+) in negative electrospray ionization mode (Table II, source parameters).


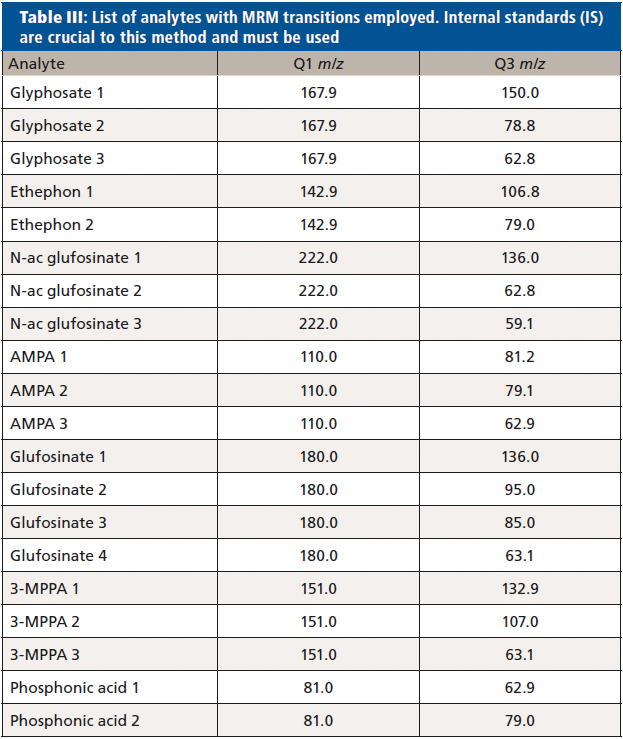
At least two multiple reaction monitoring (MRM) transitions were optimized for each analyte, in order to quantify and confirm their concentration in all samples (Table III, analytes with MRM transitions employed).


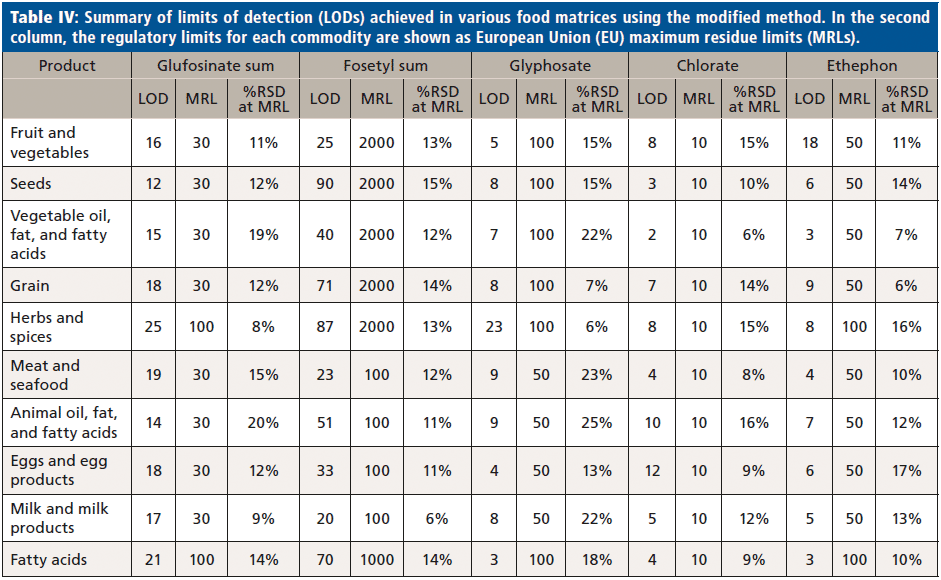
Data were acquired using Analyst 1.6.3 (Sciex) and processed for quantification and confirmation with reference to internal standards (IS), using MultiQuant 3.0.2 software (Sciex) (Table IV, summary for limits of detection).


Results and Discussion
Results
The stability and reproducibility of the method, in terms of retention time, peak width, peak area, and tailing factor, were found to be excellent, and over one thousand food samples from a variety of commodities were analyzed without system maintenance (Figure 1, reproducibility data for glyphosate internal standard [IS]).


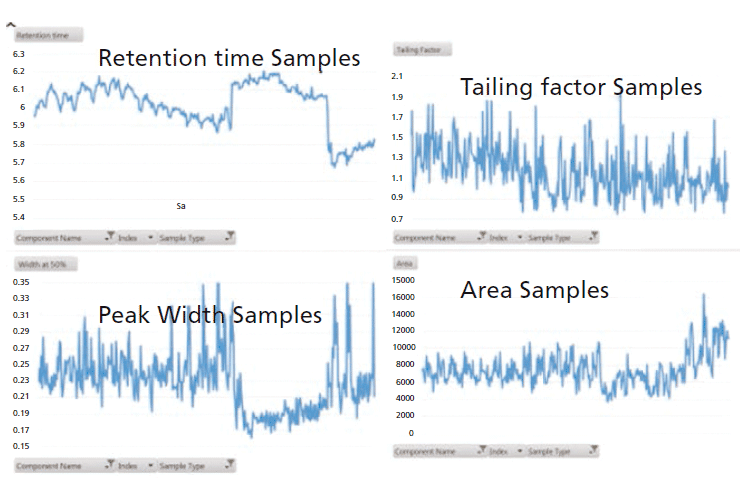
Figure 1: Reproducibility data for glyphosate internal standard (IS). The modified method for food samples tested over one hundred injections of extracts from the variety of food matrices presented in Table I.
The sensitivity of the method was demonstrated by considerably lower limit of detection (LOD) for every analyte and commodity combination than is required in most cases (Figure 3, example chromatography from surface water samples). Regulations are not expected to change significantly within the lifetime of the current generation of the most sensitive MS instruments. The linearity of the calibration curve was established in the concentration range of 15.6 to 1000 ng/L of glyphosate. By plotting the ratio of the peak area of the standard to that of the IS against the ratio of their concentrations at seven calibration levels, calibration curves were obtained (Figure 2, method sensitivity and linearity of glyphosate).


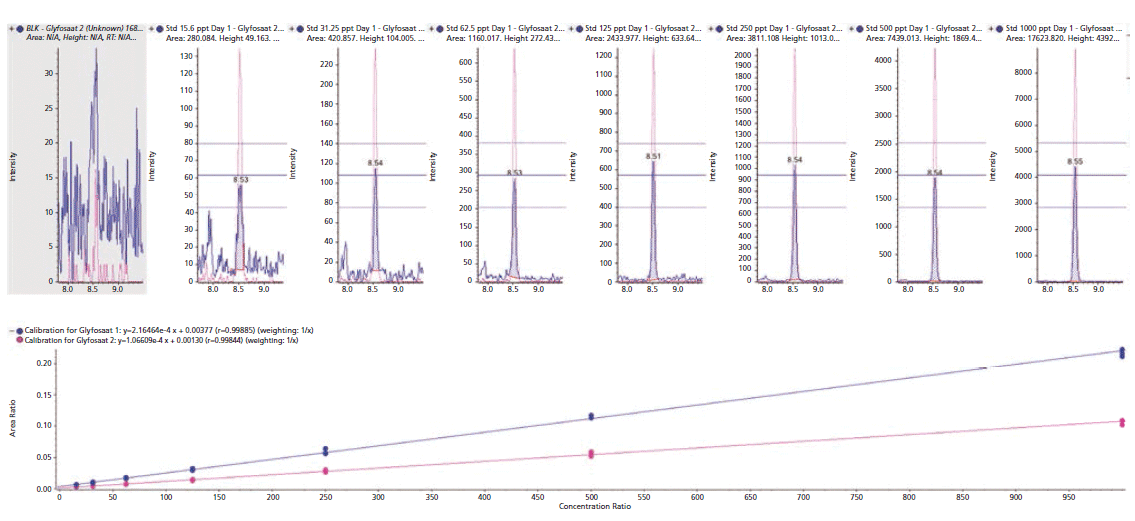
Figure 2: Method sensitivity and linearity of glyphosate. Several measures of reproducibility based on the glyphosate internal standard (IS). Calibration standards were achieved using the modified method for water samples. Ion ratios were all within the specified ±20% tolerance.
Discussion
The method described is highly robust and sensitive, achieving the target limits of detection required to meet current and proposed regulations. The separation has also been found to minimize any matrix interferences in all but the most complex samples. Through incorporation of this high throughput method into the workflows of food testing laboratories, complex sample preparation can be eliminated, running costs can be kept low, and valuable time can be saved.


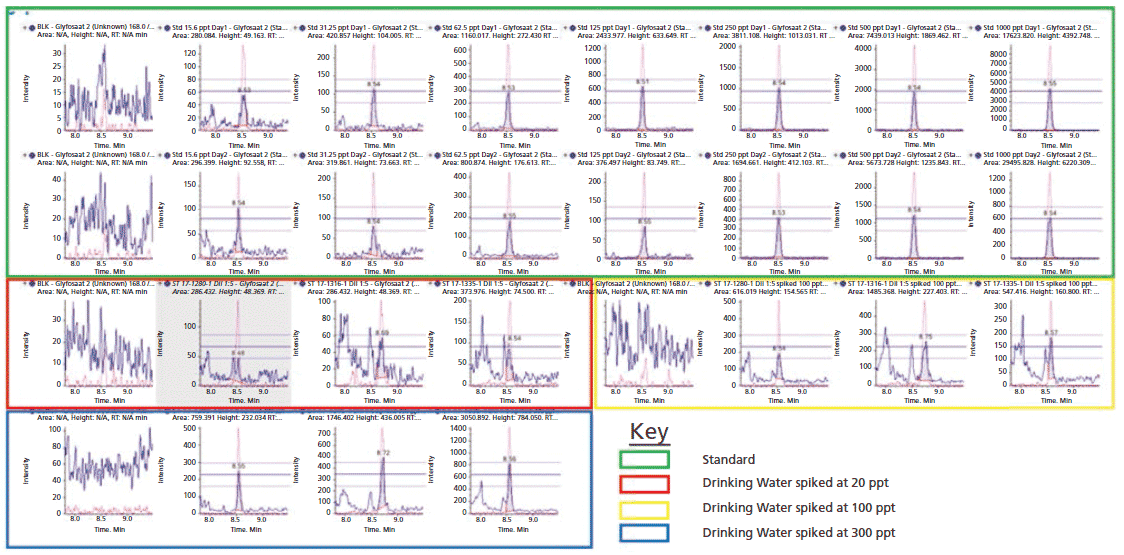
Figure 3: Example chromatography from surface water samples using the modified water method. The graphs highlight that even at concentrations five times (5x) lower than the European Union (EU) water regulations requirement, the presence of glyphosate in real environmental samples was detected and confirmed by the ion ratio between primary and secondary multiple reaction monitoring (MRM) transitions.
Chromatographic performance using these modified methods for food and water samples achieved good separation between the analytes and from matrix interferences, and excellent repeatability in terms of peak profile and retention time. This cleanup is performed on all samples, not only for food from animal origin. The ion source was clean even after more than one hundred injections.
In order to maintain peak shape and retention times, various chromatographic methods were investigated. Ion chromatography gave the most promising results, but the resulting eluents were not compatible with electrospray ionization sources and required a suppressor, which is detrimental to peak width. The need for a suppressor was removed by the use of ammonium buffers, a deviation from traditional LC buffers that enables detection of polar pesticides by MS/MS. The setup in this study allowed switching between polar and normal pesticide (reverse phase) suites without changing inlet, giving sharper, more intense peaks, and shorter run times. In busy food testing labs that primarily work with reversed-phase LC, this setup saves valuable time, eliminating the need to change inlet systems between analyses.
Food samples analyzed using the modified method for QuPPe extracts achieved unambiguous separation between the analytes as well as excellent repeatability in terms of peak profile and retention times. Analytes were also well separated from matrix interferences in most of the foods tested. The EU maximum residue limits (MRLs) for pesticides in food commodities vary widely, depending on the pesticide and commodity in question. Although some matrix interference was present in the most complex food samples, LODs in both food and water samples were well below the concentration limits laid out in EU regulation. Over one thousand food samples from a variety of commodities were analyzed without maintenance of the system, and the stability in terms of retention time, peak width, peak area, and tailing factor was found to be excellent.
This method largely eliminated matrix interferences in food sample extracts, further enhancing the reproducibility and robustness of the method. However, in the most complex food matrices, MRM ion ratios were observed outside of the standard ±20% tolerance. In future work, the QTRAP will be used to confirm positive results by their full scan MS/MS spectra, and the sample cleanup procedure will be further developed in order to remove background interferences.
It is commonly known that LC–MS/MS, especially when working in ESI mode, is susceptible to matrix effects, affecting quantitative accuracy. This new methodology overcomes the necessity of pesticide derivatization. This was achieved by combining an ion chromatography separation with the high sensitivity of the mass spectrometer for analyte detection. Analytes are successfully separated using an LC column in a method switching reversed-phase system, with MS-amenable mobile phases at around pH 9. All analytes are well retained with reproducible retention times, allowing detection of the majority of background components. The method meets the Directorate-General for Health and Food Safety (DG SANTE) requirements of reproducibility (<20%) and recovery (80 to 110%), and the limit of detection (LOD) of the method is below 0.01 mg/kg. Excellent long-term stability and robustness were achieved throughout the validation of this method for food samples extracted by the QuPPe procedure.
Conclusion
This modified LC–MS/MS analysis offers the possibility to analyze multiple polar pesticides in a single injection without derivatization. The ion chromatography based approach uses ammonium buffers, a deviation from traditional LC buffers that enables detection of polar pesticides by MS/MS. A sufficiently sensitive mass spectrometer allows the analysis to be performed without the need for an ion suppressor using a standard reversed-phase LC based system. This eliminates the need to change inlets between typical pesticide analyses.
By removing the need for derivatization, sample preparation is greatly simplified, and the time required to analyze polar pesticides is reduced. As an additional advantage, the column lifetime is well over one thousand matrix samples, allowing high-throughput polar pesticide analysis with less system maintenance. This method was found to be considerably more robust and sensitive than other approaches, and has achieved the target limit of detection required to meet existing and proposed future regulations of glyphosate and similar polar pesticides.
References
(1) M. Krüger, P. Schledron, W; Schrödl, H.W. Hoppe, W. Lutz, and A.A. Shehata, J. Environ. Anal. Toxicol. 4, 210 (2014). doi: 10.4172/2161-0525.1000210.
(2) W. Broer, U. Chiuminato, J. Stahl-Zeng, D. McMillan and P. Taylor, "A Robust and Sensitive Method for the Direct Analysis of Polar Pesticides in Food and Environmental Samples Without Derivatization" (Nofalab Laboratories, Schiedam, the Netherlands; Sciex, Darmstadt, Denmark; Sciex, Warrington, United Kingdom, 2018).
(3) M. Anastassiades, D.I. Kolberg, A. Benkenstein, E. Eichhorn, S. Zechmann, D. Mack, C. Wildgrube, I. Sigalov, D. Dörk, and A. Barth, "Quick Method for the Analysis of numerous Highly Polar Pesticides in Foods of Plant Origin via LC–MS/MS involving Simultaneous Extraction with Methanol (QuPPe-Method)" Version 9.3 (EU Reference Laboratory for Pesticides Requiring Single Residue Methods [EURL-SRM], Fellbach, Germany, August 2017).
Wim Broer and Ugo Chiuminato are with Nofalab Laboratories in Schiedam, the Netherlands. Jianru Stahl-Zeng is with Sciex in Darmstadt, Denmark. Daniel McMillan and Phil Taylor is with Sciex in Warrington, UK. Direct correspondence to: phillip.taylor@sciex.com.















New Study Reveals Insights into Phenol’s Behavior in Ice
April 16th 2025A new study published in Spectrochimica Acta Part A by Dominik Heger and colleagues at Masaryk University reveals that phenol's photophysical properties change significantly when frozen, potentially enabling its breakdown by sunlight in icy environments.




