Spectroscopy Is Applied Quantum Mechanics, Part III: Introduction to Quantum Mechanics
Columnist David W. Ball discusses the pioneering work of Erwin Schrodinger, whose work on wave mechanics forms the basis of the modern understanding of subatomic behavior.



In Part I of this series (1), I laid the groundwork for why the scientific understanding of nature in the late 19th century was found wanting: it could not explain a variety of phenomena that scientists were examining. (One of these phenomena was spectroscopy itself!) In Part II, I discussed contributions of Planck, Einstein, Bohr, de Broglie, and others who began chipping away at the façade of "Classical Science" (2). In this installment, we will discuss the pioneering work of Erwin Schrödinger, whose work on wave mechanics forms the basis of the modern understanding of subatomic behavior. Schrödinger's work is mathematically equivalent to Werner Heisenberg, Max Born, and Pascual Jordan's matrix mechanics, which has been argued as a more mathematically pure and proper form of quantum mechanics. However, most practicing spectroscopists are likely more familiar with the Schrödinger version, which is the one that will be discussed here. Note that Schrödinger published his initial work in January 1926. It took just over 25 years, from Planck's quantum theory in December 1900 to Schrödinger's work in January 1926, for the scientific community to unravel the nature of subatomic behavior. Such is the pace of science. Finally, in starting this column, I am assuming that you've read the first two installments of this series. If you haven't, you can find them archived at www.spectroscopymag.com; click on "The Baseline" link on the left of the page.
Although Max Born, Werner Heisenberg, and Pascual Jordan had published their work on matrix mechanics earlier (3,4), Erwin Schrödinger, an Austrian physicist (Figure 1), published a paper in Annalen der Physik (5) detailing another approach: an eigenvalue approach.



David W. Ball
The paper, whose title is commonly translated into English as "Quantization as an Eigenvalue Problem," introduced in its original form a second-order differential equation that had to be solved to explain the nature of the hydrogen atom. Schrödinger originally wrote the equation as


where ψ is some function, m is the mass of the electron, e is the charge of the electron, r is the radial distance between the nucleus and the electron, and E is the energy of the system. K is a constant, and must have units of action; later in the paper, Schrödinger stated that K had to equal Planck's constant divided by 2π (h/2π, now symbolized by ħ). In this equation, ∇2 is the three-dimensional second-derivative operator, so the expression is actually a second-order differential equation. Schrödinger demonstrated a solution to this differential equation that eventually produced a predicted energy of the hydrogen atom, which was exactly the same expression predicted by Bohr in his (albeit limited) theory of the hydrogen atom:


in which Schrödinger defined the integer l as his "principal quantum number." In later papers during that same year, Schrödinger presented solutions to similar differential equations based upon a harmonic oscillator, a particle constrained to a circle, and a particle constrained to a sphere. He also demonstrated that the matrix mechanics of Heisenberg and Born were mathematically equivalent and developed the concept of perturbation theory and successfully used it to explain the Stark effect (the shifting of spectral lines due to an applied electric field). Several other papers in 1926 and early 1927 cemented Schrödinger's role in the development of quantum mechanics, material from which is an important part of undergraduate physics and chemistry instruction. (An English translation of Schrödinger's nine papers of this period is available [6].)



Figure 1
The description of quantum mechanics that follows has been refined into more modern terms. In particular, note that quantum mechanics is based upon several statements called postulates, which are propositions that are not proven but are assumed to be correct. This might be unsettling to some people, but remember, high school–level geometry ultimately is based upon postulates, assumed but not proven! (Any two points can be joined by a straight line; a straight line segment can be extended indefinitely in a straight line; a circle can be drawn from a line segment using that segment as a radius; all right angles are congruent; and so forth.) What we ultimately are after are the conclusions that these postulates lead us to. If those conclusions agree with nature, we have some assurance that the postulates themselves are correct.
The Postulates of Quantum Mechanics
The postulates of quantum mechanics vary in number, depending upon the source. Rather than try to construct an exhaustive list here, I will introduce only those postulates that are necessary for our introduction to quantum mechanics. Readers are directed to any good textbook on quantum mechanics for a more complete treatment.
Postulate 1: The state of a system is described by a mathematical function called a wavefunction, represented by the lowercase Greek letter psi, ψ. Proper wavefunctions must be single-valued, bound (that is, noninfinite), continuous, and differentiable. All information that can be determined about the system must be obtained from the wavefunction.
This postulate gives the wavefunction a central role in quantum mechanics. Essentially, if you know the wavefunction of a system, you know everything there is to know, or that can be known, about that system. The four requirements of proper wavefunctions — single-valued, continuous, bound, differentiable — simply ensure that proper wavefunctions are well-behaved functions. Figure 2 shows some plots that represent proper and improper wavefunctions.


Figure 2
How do we get information from the wavefunction? The next postulate addresses that.
Postulate 2: For any observable, there exists an associated operator Ô. The only values of the observable that will be measured will be the constant K generated when the operator operates on the wavefunction and generates that constant K times the original wavefunction:


This type of equation is called an eigenvalue equation; ψ is said to be an eigenfunction of the operator Ô with an eigenvalue of K. Resist the urge to cancel ψ out of both sides of this equation, as if it were present in a multiplication operation on both sides — operating on the wavefunction with an operator is not the same as multiplying. (Many common operators, for example, contain derivatives.) The only values that will be observed will correspond to eigenvalues, and if an operator and a wavefunction do not participate in an eigenvalue equation, exact values of the observable cannot be observed. Quantum-mechanical operators are based upon the assumed forms of the position operator as x' = x• (that is, multiplying the wavefunction by the variable x) and the x-dimensional momentum operator as


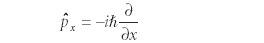
where ħ is Planck's constant divided by 2π. (Similar definitions apply for the y and z dimensions.)
If an operator and a wavefunction do not participate in an eigenvalue equation, we still can learn something about the observable associated with that operator. The average value, or expectation value, of an observable is defined in the next postulate.
Postulate 3: The average, or expectation, value of an observable, is given by the expression


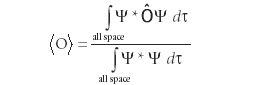
where Ô is the operator related to the observable of interest, ψ* is the complex conjugate of the wavefunction ψ, and dτ is the appropriate infinitesimal for the dimension(s) of the integrals.
Thus, the postulates of quantum mechanics allow us to find exact values of observables (but only if they come from eigenvalue equations) or average values (if you can solve the integrals listed earlier, which can always be done — at least, in theory).
So far, we have not restricted the wavefunction to being anything but a well-behaved function (finite, continuous, differentiable, single-valued). To be sure, we postulate that exact observables must come from eigenvalue equations, but we have not elevated any one eigenvalue equation to "required" status. This changes with the next postulate.
Postulate 4: Wavefunctions can be any function, but they must be eigenfunctions of the total energy operator, called the Hamiltonian operator. The one-dimensional form of the total energy eigenvalue equation is


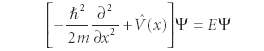
The function


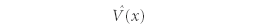
is a potential energy operator that describes the potential energy of the system of interest. The eigenvalue equation in Postulate 4 is called the time-independent Schrödinger equation and is the central equation of quantum mechanics. In understanding any atomic-scale system, we must find wavefunctions that satisfy this particular eigenvalue equation. Note that this is the same equation given in the Introduction, earlier.
The time-independent Schrödinger equation is actually a simplification of an expression called the time-dependent Schrödinger equation, which is a more fundamental form of the equation found in Postulate 4. Most day-to-day spectroscopists would have the most exposure to the time-independent form given previously.
There are other postulates — one called the completeness postulate, and several others. However, these are the basic ones that define what quantum mechanics is.
But it gets worse . . .
Uncertainty and Born's Interpretation
In operator algebra, the commutator


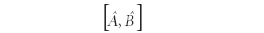
is defined as


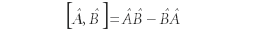
In regular multiplication, the difference in this definition would be zero, because multiplication of real and complex numbers is commutative. However, this is not the case with operators. This can be demonstrated easily by multiplying two 2×2 matrices:


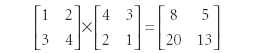
But:


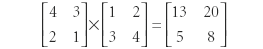
Granted, the matrix elements in the final products are the same, but because they are in different positions, they are actually different matrices. The same can be true with operators. For some operators, it does not matter what the order of operation is. If this is the case, the value of the commutator is zero and the operators are said to commute. If the order of operation does matter, the value of the commutator is nonzero and the operators do not commute.
In 1926, Werner Heisenberg demonstrated that if one defines an error in the expectation value of position Δx as


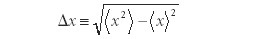
and the error in the expectation value of momentum Δpx as


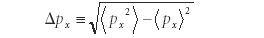
then, using a mathematical relationship of operators called the Cauchy–Schwarz inequality (a relationship known since the early 1800s but which is beyond the scope of this column; interested readers are urged to research the topic further), the following relationship between Δx and Δpx had to exist:

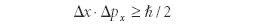
This relationship is called the Heisenberg Uncertainty Principle. Notice that this is not a postulate of quantum mechanics; rather, it is a logical consequence of quantum mechanics' use of operator mathematics. The Uncertainty Principle demonstrates that it is impossible to know the position and the momentum of a particle to within certain fundamental limits, those limits being determined by Planck's constant. The Uncertainty Principle might not bother us at the macroscopic level, but at the level of individual electrons, it is devastating to our ability to understand exactly where an electron is. The Uncertainty Principle says, in effect, we can't.
How does one reconcile the Uncertainty Principle with the idea that the wavefunction gives the state of a system? Probably the best way to understand the wavefunction in light of the Uncertainty Principle was enunciated by Max Born in 1927. The Born interpretation, or Copenhagen interpretation (so-called because Niels Bohr set up a working group of physicists to develop the new quantum mechanics in Copenhagen, Denmark) says that the wavefunction should not be interpreted as indicating exactly where a particle is; rather, it is related to the probability that a particle is in a particular region given by the set of boundaries {a, b} at any given time. The probability is given by


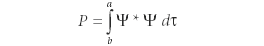
where again ψ* is the complex conjugate of the wavefunction ψ and dt is the appropriate infinitesimal for the coordinates of the region. If the region is the entire space of the system, the probability had to be 100% or, in decimal terms, 1, so the previous equation leads to another requirement of wavefunctions: that numerically, the following integral must be satisfied:


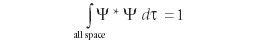
When this condition is satisfied, the wavefunction is said to be normalized. In any case, the Born interpretation reduced the wavefunction to indicating a probability, not a certainty.
Many scientists had (and still have) a problem with this. Some people could not accept that the behavior of matter is, at its ultimate, determined by probabilities. A strong history of determinism, of specific causes and effects, pervaded classical science, and to reduce the behavior of atoms and subatomic particles to just probabilities destroyed that concept. As distinguished a scientist as Albert Einstein was troubled by this, apparently saying "Ich, auf jeden Fall, morgens überzeugte, daß er nicht Würfel wirft." ("I, at any rate, am convinced that He [God] does not play dice.") To which Niels Bohr, apparently more accepting of the strange conclusions of quantum mechanics, replied, "Don't tell God what to do."
Although radical at the time, quantum mechanics has been accepted as the best theory for explaining behavior at the atomic and subatomic levels. In the next installment of this column, we will consider some ideal systems using quantum mechanics and discuss some tools of quantum mechanics that we use to explain spectroscopy.
David W. Ball is a professor of chemistry at Cleveland State University in Ohio. Many of his "Baseline" columns have been reprinted in book form by SPIE Press as The Basics of Spectroscopy, available through the SPIE Web Bookstore at www.spie.org. His most recent book, Field Guide to Spectroscopy (published in May 2006), is available from SPIE Press. He can be reached at d.ball@csuohio.edu his website is academic.csuohio.edu/ball.
Erratum
In the last installment of this column (December 2007, p. 92), the Rydberg constant was incorrectly stated as 1.09737 × 10–7. It should be 1.09737 × 107. Many thanks to Dr. Stephen E. Mitchell of the Remote Sensing Laboratory, U.S. Department of Energy, for kindly pointing this out.
References
(1) D.W. Ball, Spectroscopy 22(12), 91–94 (2007).
(2) D.W. Ball, Spectroscopy 23(1), 18–22 (2008).
(3) M. Born and P. Jordan, Z. Phys. A 34, 858–888 (1925).
(4) M. Born, W. Heisenberg, and P. Jordan, Z. Phys. A 35, 557–615 (1926).
(5) E. Schrödinger, Ann. Phys. 79, 361–376 (1926).
(6) E. Schrödinger, Collected Papers on Wave Mechanics, English translation. 2nd ed. (Chelsea Publishing Company, New York, 1978).
















Smarter Sensors, Cleaner Earth Using AI and IoT for Pollution Monitoring
April 22nd 2025A global research team has detailed how smart sensors, artificial intelligence (AI), machine learning, and Internet of Things (IoT) technologies are transforming the detection and management of environmental pollutants. Their comprehensive review highlights how spectroscopy and sensor networks are now key tools in real-time pollution tracking.



New AI Strategy for Mycotoxin Detection in Cereal Grains
April 21st 2025Researchers from Jiangsu University and Zhejiang University of Water Resources and Electric Power have developed a transfer learning approach that significantly enhances the accuracy and adaptability of NIR spectroscopy models for detecting mycotoxins in cereals.

Karl Norris: A Pioneer in Optical Measurements and Near-Infrared Spectroscopy, Part II
April 21st 2025In this two-part "Icons of Spectroscopy" column, executive editor Jerome Workman Jr. details how Karl H. Norris has impacted the analysis of food, agricultural products, and pharmaceuticals over six decades. His pioneering work in optical analysis methods including his development and refinement of near-infrared spectroscopy, has transformed analysis technology. In this Part II article of a two-part series, we summarize Norris’ foundational publications in NIR, his patents, achievements, and legacy.

