What's New with the FDA's Data Integrity Guidance?
In April 2016, the FDA issued a draft guidance for industry on data integrity and compliance with cGMP. What does this mean for the laboratory?
In April 2016, the United States Food and Drug Administration (FDA) issued a draft guidance for industry called “Data Integrity and Compliance with cGMP (current good manufacturing practice).” What does this mean for a regulated laboratory?
Data integrity is currently the hottest topic in regulated good practice (GxP) laboratories as readers of this column will know. So far, this year has seen the publication of the final version of the World Health Organization (WHO) guidance on “Good Data and Records Management Practices” (1) as well as a draft from the United States Food and Drug Administration (FDA) guidance for industry entitled “Data Integrity and Compliance with cGMP (current Good Manufacturing Practice)” (2). These publications now join the United Kingdom’s Medicines and Healthcare Products Regulatory Agency (MHRA) data integrity guidance issued in March 2015 (3). As this column was being written, the MHRA issued an update on their data integrity guidance for consultation where the overall scope is expanded to GxP (4). Not to be outdone, the European Medicines Agency (EMA) (5) and Pharmaceutical Inspection Convention/Pharmaceutical Inspection Cooperation Scheme (PIC/S) (6) also issued data integrity guidance in August as this column went to press.
A veritable cornucopia of data integrity regulatory advice and guidance is upon us! Or perhaps another perspective is a tsunami of advice from the regulators? This abundance of regulatory guidance is shown graphically in Figure 1, and you can see that the FDA has more guidance and advice for data integrity than any other regulatory source:
- Guide to the Inspection of Pharmaceutical Quality Control Laboratories (7)
- Compliance Program Guide (CPG) 7346.832 for Pre-Approval Inspections (PAI) (8) Level 2 guidance on the FDA website (9)
- Draft Guidance for Industry, Data Integrity, and Compliance with cGMP (2)


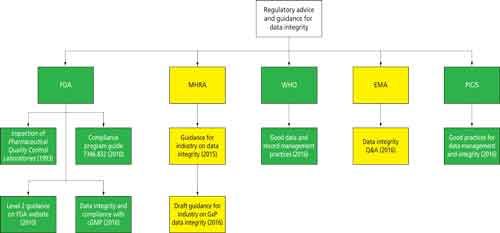
Figure 1: Data integrity guidance from regulatory sources.
CLICK FIGURE TO ENLARGE
Although “Guide to Inspections of QC Labs” (7) is more than 20 years old, it is still worth reading because most laboratories have not changed their working practices greatly in the intervening time since its publication. For example, it includes the statement, “The authority to delete files and override computer systems should be thoroughly examined.”
For the purposes of this column, we focus our discussion on the FDA guidance document on data integrity although the discussion will also bring up some of the other FDA guidances in Figure 1 as well as some other documents issued by the agency. This column installment is not a comprehensive review of the data integrity guidance (2)-the focus here is on those sections that impact on the regulated good manufacturing practice (GMP) laboratory. The principles discussed here can be extended to any regulated or quality laboratory.
Overview of the FDA Guidance
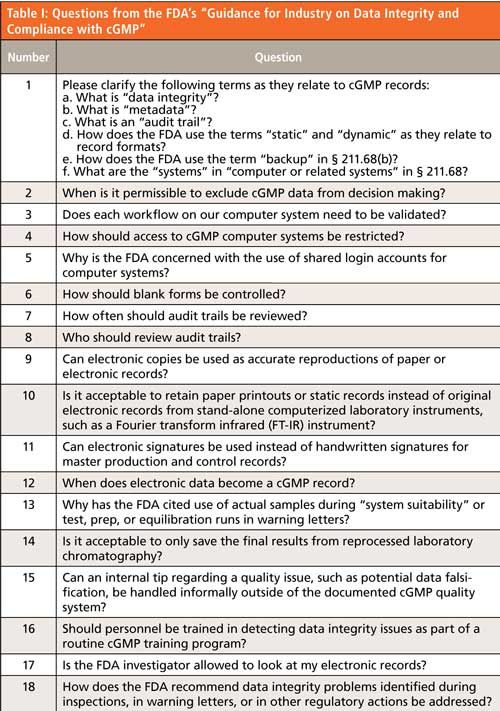
The FDA guidance is unlike those from the WHO and MHRA guidance documents (1,3) in that it is presented in the format of 18 questions and answers, as shown in Table I. The FDA document does not have the more encompassing scope of the MHRA and WHO guidance documents that consider topics such as data governance, the role of management, and the extension of data integrity to an organization’s suppliers. Instead, the FDA guidance is complementary and is entirely focused on the interpretation of the 21 CFR 211 regulations for current GMP (cGMP), specifically to ensure the integrity of data generated in pharmaceutical manufacturing (10). The problem with US regulations, unlike those in the European Union, is that (with one exception) they have not been updated since 1978. As such there is no explicit reference that is specific for ensuring the integrity of laboratory data-it is the interpretation of the regulations that is key. As a result, there are multiple references to the different sections of 21 CFR 211 to support the 18 questions. Of particular interest is question 1e, which illustrates the “current” in cGMP (2). Backup is now interpreted by the FDA as long-term archives for records retention rather than simply creating a copy of records on tape or disk for disaster recovery purposes.


Question 1d: Static Versus Dynamic Data
Question 1d talks about static and dynamic data, which are perhaps not the best of terms to use in describing data. Static data are typically discrete values such as temperature and pH that cannot be interpreted or as the guidance mentions a paper printout or image. In contrast, dynamic data requires human interpretation or processing, such as with chromatography or spectroscopic data files, and these types of data are of major concern to the FDA and other regulators for manipulating data and testing to pass. “Processable” data might be a better term because we typically process our data in the laboratory.
Questions 4 and 5: Access to Computerized Systems
In short, there must not be any generic or shared log-on accounts for access to a computerized system because each person must be uniquely identified and their actions within a system tracked and audit trailed. User types need to be established that separate administrator privileges from those involved with generating, processing, and reviewing data. Ideally, an independent function, typically the IT department, needs to control the administration rather than the laboratory. However, with standalone systems this independent control may be impossible to achieve and therefore as noted in the MHRA data integrity guidance (3) an alternative option may be for a laboratory user to have two user types. The first would be as an administrator with no user access rights and the second as a user with no administrator privileges.
The FDA guidance also recommends maintaining a list of authorized individuals with their access privileges. This list should cover both current and historical users of a system. In case you think this recommendation is an FDA rabbit out of the hat, it has been the stated agency position since 2007. It is contained in the guidance for industry on “Computerized Systems Used in Clinical Investigations” (11) under “Recommendations, Section E” on external security safeguards. It states, “You should maintain a cumulative record that indicates, for any point in time, the names of authorized personnel, their titles, and a description of their access privileges.”
This is good advice because it allows quality assurance, auditors, or inspectors to see at any point in time the access privileges that any individual has had for a system, including those of trainees, analysts, and supervisors.
Questions 12 and 2: When Do Data Become a GMP Record and Can I Exclude GMP Data?
Now we come to probably the most contentious part of the FDA guidance: question 12. At the beginning of question 12 is a simple statement of fact, “When generated to satisfy a GMP requirement, all data become a GMP record.” This is simply a confirmation and restatement of the GMP requirement in 21 CFR 211.194(a) for complete data secured in the course of testing (10).
At this point can I mention the “s” word? Sssh, whisper it softly . . . spreadsheet! How many laboratories use validated spreadsheet templates only to print but not save the completed files? This recommendation means that the completed file must be saved under a unique filename and if signed or initialed there must be a linkage between the saved file and the signed printout to comply with 21 CFR 11.70 (12). Simply printing the spreadsheet to a PDF does not comply with this requirement-the original file must be saved.
The guidance then continues:
You must document at the time, or save, the data at the time of performance to create a record in compliance with GMP requirements . . . FDA expects processes to be designed so that quality data required to be created and maintained cannot be modified.
For example, chromatograms should be sent to long-term storage (archiving or permanent record) upon run completion instead of at the end of a day’s runs.
There is a slight problem here: if a spectroscopy or chromatography file cannot be modified how can we interpret it? Perhaps what is meant is that the data or the computer file created cannot be changed, but the data contained within it can be interpreted?
The requirement about removing chromatograms immediately to long-term storage after each run borders on the silly side and does not reflect reality. I appreciate what the agency is trying achieve-to ensure that electronic records generated by data systems of all types are protected, especially for standalone systems that do not have databases. This actually reflects on how instrument suppliers design and laboratories select data systems that are substandard for regulated laboratories. To put this into perspective, virtually all software currently in use in laboratories was designed before the current focus on data integrity. For too long laboratories have accepted spectroscopic applications running on standalone workstations that generate data files that are stored in directories in the operating system. Quite simply, these systems are not fit for purpose. To illustrate this point, McDowall and Burgess recently wrote a series of four papers describing the ideal chromatography data system (CDS) for regulated GxP laboratories in LCGC North America (13–16). Although focused on chromatography data systems, most of the principles and recommendations outlined in these papers are also applicable to spectroscopic data systems. In the part on system architecture (14), the point was made that standalone workstations and using directories in the operating system for file storage were not fit for purpose. Instead data must be acquired directly to network storage and that all data systems must use a database. In doing so, the intent of the FDA’s requirement would be met but in a more practical way. Correctly designed data systems are the main way laboratories will comply with the protection of records other than implementing a scientific data management system (SDMS).
Question 12 progresses through the next statement in the document for paper records, which must have been copied verbatim from a GxP documentation class 101. However, as judging from the citations in many warning letters there appears to be an overabundance of idiots working in regulated laboratories so that the following needs to be repeated for temporary scraps of paper: “It is not acceptable to record data on pieces of paper that will be discarded after transcription into a permanent laboratory notebook.”
There then follows the electronic equivalent, which is more contentious:
Similarly, it is not acceptable to store data electronically in temporary storage, in a manner that allows for manipulation, before creating a permanent record. Electronic data that are automatically saved into temporary memory do not meet cGMP documentation or retention requirements.
This section has probably caused more discussion than any other in the whole draft data integrity guidance. Obviously the agency does not want people taking a file then interpreting it multiple times without saving it, because this is testing-or rather over-interpreting-into compliance.
I don’t believe that the FDA wants keystroke loggers on all systems used for regulated analysis. The process in all data systems should be to save the data first and monitor the interpretation. However, this is where science and compliance meet, often in a head-on collision. The controls required, especially technical ones rather than procedures, need to be designed and implemented. In some systems, especially those close to the research end of research and development (R&D), may be designed with minimal regulatory compliance controls. There needs to be a fundamental rethinking by software suppliers about how their software is designed and operated in compliance with the applicable regulations. However, this process will take time as anyone will know who has seen some of the shambolic attempts to produce technically compliant Part 11 applications or are waiting now for a function to document second person review of audit trail entries for Annex 11. This step is the scientific equivalent of Waiting for Godot. For readers who have not seen the play, you sit through two hours and Godot never appears.
It should be noted that although this “Focus on Quality” installment is about the FDA’s “Data Integrity Guidance,” the recent UK MHRA guidance includes this statement, “. . . it is expected that GMP facilities should upgrade to an audit trailed system by the end of 2017.”
It is nice to know that optimism still exists in the UK. Given the lead time for suppliers to implement these controls in software and the lack of laboratory users to press suppliers for these features means that this deadline will come and go with no change. This is the situation with small-molecule chemistry software applications. The situation with biotechnology software is even worse-the regulatory compliance equivalent of the Wild West as it is closer to research.
Question 2 discusses if GMP data can be excluded from decision making. It notes that, “any data created as part of a GMP record must be evaluated by the quality unit as part of release criteria and maintained for GMP purposes. Electronic data generated to fulfill GMP requirements should include relevant metadata.”
The answer is that data (paper, hybrid, or electronic) can only be excluded if there is a justified and documented scientific rationale-for example, an out-of-specification result following a laboratory investigation. The corollary is that data should not be deleted, even if it is excluded, because it is part of complete data collected in the course of testing under 21 CFR 211.194(a) (10). In this context, there has been an interesting publication recently entitled “18 Reasons Spelt Out by Peter J. Werth to Delete Analytical Data” (15), which range from incorrect sample preparation to plumbing problems and leaks. Perhaps this publication is an isolated view from the owner of an active pharmaceutical ingredient company, but senior management need to be aware of the issues of data integrity and comply with regulations. If they choose to ignore this advice, then as some children’s TV shows warn: don’t do this at home. Do not follow this advice and delete data. From the answers in questions 2 and 12 deletion of GMP data without a documented justification (2) can seriously damage your company’s wealth because of noncompliance with GMP.
Question 3: Does Each Workflow on a Computer System Need Validating?
Yes, is the answer in the FDA guidance (2). If a workflow is configured or customized then it needs to be specified, built, or configured in the software and then tested for intended use. So far, so good, and I have no problem with this approach.
However, what if you have standard workflows in a system that you don’t use and can’t turn them off? As the question is written and answered, the data integrity guidance implies that they all need to be validated, which is a compliance overhead and not in the spirit of a risk-based approach. There is also a clash of FDA guidance documents. There is the small matter of the 2002 “General Principles of Software Validation,” which states in section 6.1 (18):
For example, a (device) manufacturer who chooses not to use all the vendor-supplied capabilities of the software only needs to validate those functions that will be used and for which the (device) manufacturer is dependent upon the software results as part of production or the quality system.
Or put simply, only validate those software functions you use. Note that I have added the parentheses in the quotation above because this guidance was written for medical devices. As currently written, there is a potential clash of guidance from the FDA and hopefully in the final version of the data integrity guidance there will be clarification to align the data integrity and software validation guidance documents so that there is no conflict.
Question 6: Control of Blank Forms
At the back of many procedures in regulated laboratories are blank forms designed to ensure compliance with the work contained in the standard operating procedure (SOP) and to collect the required data. Question 6 of the FDA guidance raises the question of how these blank forms should be controlled (2). The FDA wants each copy of such forms to be uniquely numbered and accounted for. Does this sound like the agency is making up rules as they go along? Actually, it is not and is simply a restatement of their position from the 1993 guidance on “Inspection of QC Laboratories” (7) as noted in section 13 on documentation: “We expect raw laboratory data to be maintained in bound (not loose or scrap sheets of paper) books or on analytical sheets for which there is accountability, such as prenumbered sheets.”
The requirement for control of blank forms has also been presented in the recent MHRA and WHO guidance documents (1,3). Furthermore, this topic is also covered in some detail in the recently published EMA and PIC/S data integrity guidance documents (5,6). The rationale for this approach is that uncontrolled blank forms present an opportunity for data falsification or testing into compliance. This is an important area that is the subject of my next “Questions of Quality” column in Spectroscopy’s sister publication, LCGC Europe (19). The bottom line is that there is now an administrative burden on analytical laboratories that will, sooner or later, force organizations to move to electronic working to reduce the overhead and automate this aspect of regulatory compliance.
Importance of the Second Person Review
A problem with the FDA data integrity guidance is that there is only a focus on audit trail review with questions 7 and 8 and with question 16 on the need for personnel be trained to detect data integrity issues (2). In my view, the draft guidance misses the point and an opportunity. If we are serious about data integrity and compliance with the regulations, surely the focus both here and in our laboratories should be on a series of questions covering the second person review of analytical data. As currently written, the guidance is scratching the surface with simply a focus on a computerized system audit trail (if used) and a suggestion that people should be trained to detect poor data management practices. Perhaps there should be questions such as
- How should I review paper records? Here the focus could be on the objective evidence available so that the second person reviewer can check that an analytical procedure has been followed. Alternatively, if we are dealing with a color or odor test, the reviewer can go to the sample to check the observation made by the tester.
- How should hybrid records be reviewed? In this case, the reviewer needs to check that the data and all metadata have been collected and data collection SOPs have been followed. The review would also include a risk based review of the audit trail entries involved with modification (and deletion if allowed) of data. This review would involve the data system. There would also be a check of any records outside of the system, such as sample preparation and input of data into the data system plus that the printout corresponds to the data in the application.
- How should electronic records and electronic signatures be reviewed? When a reviewer is working electronically, the review is based on the technical controls implemented to ensure that the workflow was followed and any calculations as well as the application is validated (20). When a reviewer is working electronically, with a validated workflow that enforces activities and validated calculations, the focus can be on the interpretation of the data and audit trail entries, simplifying the whole review. If the application has the functionality, the audit trail can identify which audit trail entries the reviewer should check. However, even in an electronic system as with a hybrid, samples are prepared outside of the system and data will be manually input, these values also need to be checked.
- How are sample preparation activities to be recorded and, more pertinently, reviewed (for example, sample dilution and preparation)? Perhaps it’s time to buy shares in video recorder companies?
These are only my suggestions, but the guidance needs to be developed further to cover this gap.
Question 10: Can I Retain Paper Printouts Rather than Electronic Records?
For simpler instruments such as an analytical balance and a pH meter, the paper printouts are the GMP record and must be retained. However, the data integrity guidance also quotes an example of a Fourier transform infrared (FT-IR) spectrometer where it is used in a GMP activity, such as identification of materials against a library. Here the electronic record is dynamic or processable, and thus paper should not be the only record because the printout may only display part of the sample spectrum. However, the argument in the draft guidance is not as well presented as the original discussion on this topic that is on the FDA’s website, which poses the following question (9): “3. How do the Part 11 regulations and ‘predicate rule requirements’ (in 21 CFR Part 211) apply to the electronic records created by computerized laboratory systems and the associated printed chromatograms that are used in drug manufacturing and testing?”
In this instance, the 2010 argument began from the premise that the FDA believed that some in industry were misinterpreting Part 11 and replying on paper printouts and keeping the underlying electronic records as the raw data from an analysis. The problem is that the question begins generally talking about “computerized laboratory systems” but by mentioning “associated printed chromatograms” brings the focus of the question to a specific analytical technique. This mention of chromatography means that spectroscopists immediately turn off because the guidance is not seen as being written for them. In reality, the focus should be on the computerized laboratory system, which would include all spectroscopic and chromatographic data systems and also, I would argue, spreadsheets. The discussion could be illustrated by FT-IR and chromatography examples, but in my view the final version of the guidance should be aimed at a wider analytical audience.
The following two sections of the GMP regulations are quoted both on the website and the guidance to support why electronic records and not paper should be the raw data:
- §211.180(d), which notes that paper printouts are not the original records or true copies of the underlying e-records
- §211.68(b), which states that paper is not a complete and exact copy of the electronic records as the latter contain more information than printouts.
A better worded discussion that melds both the current draft guidance and the website advice together would be a better approach in this area.
What About the Part 11 Scope and Application Guidance?
It’s a funny old world, isn’t it? You wait for a data integrity guidance from the FDA and then you think, how does this mesh with other existing guidance documents? We have seen that, for the most part, there is consistency in the FDA’s approach to blank forms and security, but what about the Part 11 “Scope and Application Guidance” (21)? A draft was issued in February 2003 with a 60-day comment period, and the final version was issued in September 2003. That was a lightning fast turnaround for an FDA guidance for industry. The guidance document is now 13 years old and the 21 CFR 11 regulations are nearly 20 years old. I would like to pose the following questions:
- What is the value of 21 CFR 11 regulations? It may appear that the sole purpose of 21 CFR 11 is to allow industry to use electronic signatures and software suppliers to ensure suitable technical controls are included in applications. The main means of enforcement by the FDA is via the cGMP regulations.
- Should the Part 11 guidance be retired or merged with the data integrity document? Warning letters based on 21 CFR 11 regulations have been nonexistent for the past 13 years and all citations for data integrity have involved the predicate rule (21 CFR 211) regulations.
Furthermore, the Part 11 guidance provides enforcement discretion for:
- 11.10(a) validation -This sub clause also includes the ability of systems to discern altered records, the trigger for an audit trail entry. In the light of data integrity issues, is it still wise to have this available for anyone to read and misinterpret?
- 11.10(e) audit trails-Given the current emphasis and importance of audit trails shouldn’t the enforcement discretion be lifted? The systems it refers to are probably long past their retirement date or are about to expire. Data integrity is the more important topic to focus on, and there should be statements in the final version of the data integrity guidance about how these two guidance documents interact and which takes precedence.
Summary
In this column, we looked at the elements from the draft FDA “Guidance on Data Integrity and cGMP Compliance” that have an impact on a GMP regulated laboratory. Many items are common sense and understandable, while others are less so and clash with existing guidance from the FDA. Hopefully, these unresolved issues will be addressed in the final version of the document.
Acknowledgment
I would like to thank Paul Smith for comments made in the preparation of this column.
References
- World Health Organization, WHO Technical Report Series No.996 Annex 5 Guidance on Good Data and Records Management Practices (WHO, Geneva, Switzerland, 2016).
- FDA, “Draft Guidance for Industry Data Integrity and Compliance with cGMP,” proposed, Federal Register, 2016, Silver Spring, Maryland.
- Medicines and Healthcare Products Regulatory Agency, MHRA GMP Data Integrity Definitions and Guidance for Industry, 2nd Edition (MHRA, London, England, 2015).
- Medicines and Healthcare Products Regulatory Agency, MHRA Draft GxP Data Integrity Definitions and Guidance for Industry (MHRA, London, England, 2016).
- EMA Questions and Answers: Good Manufacturing Practice: Data Integrity (2016). Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/gmp_q_a.jsp&mid=WC0b01ac058006e06c#section9.
- PIC/S PI-041 Draft Good Practices for Data Management and Integrity in Regulated GMP / GDP Environments. 2016, Pharmaceutical Inspection Convention / Pharmaceutical Inspection Co-Operation Scheme, Geneva Switzerland.
- US Food and Drug Administration, Inspection of Pharmaceutical Quality Control Laboratories (FDA, Rockville, Maryland, 1993)
- US Food and Drug Administration, Compliance Program Guidance CPG 7346.832 Pre-Approval Inspections (FDA, Silver Springs, Maryland, 2010)
- US Food and Drug Administration, FDA Questions and Answers on Current Good Manufacturing Practices, Good Guidance Practices, Level 2 Guidance-Records and Reports, 2010. Available from: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm124787.htm.
- Current Good Manufacturing Practice for Finished Pharmaceutical Products, 21 CFR clause 211 (U.S. Government Printing Office, Washington, DC, 2008).
- US Food and Drug Administration, FDA Guidance for Industry: Computerised Systems Used in Clinical Investigations (FDA, Rockville, Maryland, 2007).
- Current Good Manufacturing Practice for Finished Pharmaceutical Products, 21 CFR clause 11 (U.S. Government Publishing Office, Washington, DC, 1997).
- R.D. McDowall and C. Burgess, LCGC North Am.33(8), 554–557 (2015).
- R.D. McDowall and C. Burgess, LCGC North Am.33(10), 782–785 (2015).
- R.D. McDowall and C. Burgess, LCGC North Am.33(12), 914–917(2015).
- R.D. McDowall and C. Burgess, LCGC North Am.34(2), 144–149 (2016).
- (17) P. Werth, “18 Reasons Spelt Out by Peter J. Werth to Delete Analytical Data, Pharma Compass, 2016.
- US Food and Drug Administration, FDA Guidance for Industry General Principles of Software Validation (FDA, Rockville, Maryland, 2002).
- C. Burgess and R.D. McDowall, LCGC Europe,29(9), in press (2016).
- R.D.McDowall, LCGC Europe 29(6), 310–316 (2016)
- US Food and Drug Administration, Guidance for Industry, Part 11 Scope and Application (FDA, Rockville, Maryland, 2003).



R.D. McDowall is the Principal of McDowall Consulting and the director of R.D. McDowall Limited, as well as the editor of the “Questions of Quality” column for LCGC Europe,Spectroscopy’s sister magazine. Direct correspondence to:
SpectroscopyEdit@UBM.com














