What's New in the Proposed USP Update?
A proposed update to USP on Analytical Instrument Qualification (AIQ) has been issued for public comment. Here, we identify the changes that are proposed and their impact on the AIQ process of analytical instruments and laboratory computerized systems.
Changes have been proposed to the United States Pharmacopeia Chapter <1058> on analytical instrument qualification. Here is what you need to know about the impact of those changes on the qualification of analytical instruments and laboratory computerized systems.
The United States Pharmacopeia (USP) general chapter <1058> on analytical instrument qualification (AIQ) (1) has been effective since 2008. It outlines an approach to the qualification of analytical instruments and laboratory computerized systems based on the 4Qs model life cycle. As background for this installment let me briefly recap the history of this general chapter, which is the only general chapter in the major pharmacopeias to focus specifically on analytical instrument qualification. The European Pharmacopoeia and Japanese Pharmacopoeia do not have equivalent chapters on the subject, although in some instrument-specific general chapters there are some qualification criteria.
USP <1058> started as the conference called Analytical Instrument Validation that was organized in 2003 by the American Association of Pharmaceutical Scientists (AAPS). The first item agreed on by the conference was that the title was wrong-it should be Analytical Instrument Qualification. The rationale for this change was that we validate processes, systems, and analytical procedures and qualify equipment. In 2004, AAPS published the output of the conference as a white paper (2) that described the process of instrument qualification as the 4Qs model:
- Design qualification (DQ)
- Installation qualification (IQ)
- Operational qualification (OQ)
- Performance qualification (PQ)
The 4Qs model is the same as that published by the UK’s Pharmaceutical Analytical Science Group (PASG) in 1995 when they presented their approach to equipment qualification for the analytical laboratory (3). In addition there were several other papers published in the mid-to-late 1990s on equipment qualification (4–7).
There is a good, if somewhat simplistic, risk based classification for equipment used in a regulated laboratory in chapter <1058> (1). In essence, anything can be classified into the following groups:
- Group A: Equipment that is qualified by observation; typically items in this group have no calibration or maintenance requirements.
- Group B: Instruments that either measure or control a physical property, and any software is in the form of firmware that is implicitly or indirectly validated when the instrument is qualified.
- Group C: Systems that typically consist of an instrument controlled by software installed on a separate workstation. The software usually controls the instrument, acquires data, interprets the data, reports results, and stores the data. The instrument needs to be qualified and the software needs to be validated.
Over the past few years, I have written and commented on the content of this general chapter in various installments in this column for Spectroscopy (8–14). Like the curate’s egg (15), USP <1058> is good in parts. In January 2012, Chris Burgess and I published a stimulus to the revision process in Pharmacopeial Forum (16) that advocated an integrated approach to risk-based analytical instrument qualification and computerized system validation (AIQ-CSV) for laboratory computerized systems. This was intended to be incorporated in a revision to USP <1058>. A detailed, risk-based approach was published by us in this column in 2013 (17) in which group B instruments and group C systems were each subdivided into three subtypes. This approach allowed analytical chemists to target their validation and qualification work on the intended use of the system rather than adopt a fixed one-size-meets-all mentality. Following our feedback on industry comments from the stimulus paper, Chris and I drafted an update to <1058> in the summer of 2013. The draft was reviewed by several experts in the pharmaceutical industry and suppliers before submission to the USP expert committee. In the May–June 2015 issue of Pharmacopeial Forum (18) a proposed update to USP <1058> was published for public comment. This column reviews what has changed in this proposed version of USP <1058>.
Problems with the Current USP <1058>
As a starting point, let’s look at my critique of the current version of USP <1058> based on the “Focus on Quality” column that I wrote in November 2010 (8). Summarizing my concern with the current version of USP <1058>, the following issues are noted:
Problem 1: Users Are Responsible for DQ
The current version of the general chapter says mainly that the manufacturer is responsible for DQ. Rubbish! The user is responsible for the system as he selects and uses the instrument or system to generate regulated data.
Problem 2: The True Role of the Manufacturer Is Missing
The manufacturer or supplier is involved throughout the integrated AIQ-CSV process. This starts with the design of the instrument or system, through training to use the item, qualification services, calibration and requalification, and updates of software. This is the elephant in the room. The role of the manufacturer needs to be integrated into the process more.
Problem 3: Poor Software Validation Guidance
The implicit validation of software in Group B instruments is a good approach and was consistent with the approach advocated in Good Automated Manufacturing Practice (GAMP) versions 1–4 (19). However, consistency disappeared with the dropping of GAMP software category 2 in the current version of the GAMP guide (20). Schuessler and colleagues (21) have mapped the current GAMP software categories against the USP groups including the subcategories advocated by Burgess and McDowall (16,17). Furthermore, the Food and Drug Administration (FDA) validation reference (22) quoted in USP <1058> is intended for medical device software and not configurable software often seen in analytical laboratories.
Problem 4: Groups Can Be Misleading
There is a list of devices that are placed in the three groups; however, this can be misleading. For example, in the current version of <1058> a dissolution bath is placed in Group C. In reality, a standalone dissolution bath is a Group B instrument that only undergoes calibration and any firmware is implicitly validated through the qualification process. However, if the dissolution bath is controlled by a separate computer system, then it will be classified as a Group C item. This approach has the potential to lead to confusion by placing an instrument in either too high a group and doing too much work or in a lower group and generating a compliance gap.
What Has Changed in the Proposed <1058>?
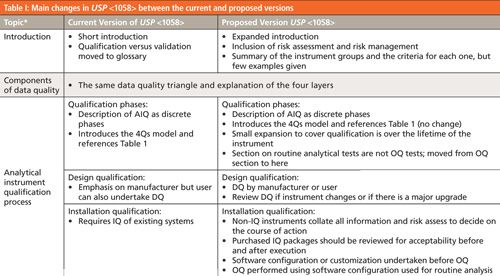
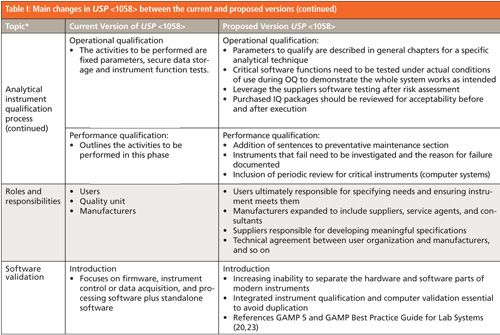
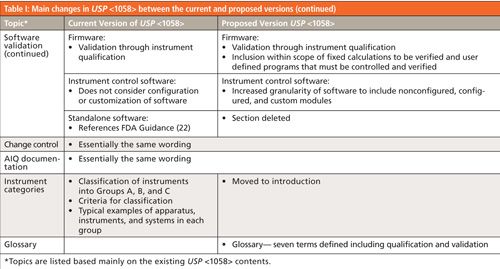
The best way to summarize the changes in the proposed version of USP <1058> is by comparing the two versions. This is done in Table I, which shows the topics in the current version of <1058> (mainly) with one or two additions from the new version of the general chapter. The center and right-hand columns summarize the content and many changes in the new and proposed versions, respectively. I should sound a note of caution here: This is a summary of changes and does not go into depth. For more detail you should read both the current and proposed versions of <1058> to glean this detailed information.






Is the Proposed USP <1058> Better?
Not all improvements to regulations and regulatory guidance may be good or even required. Therefore, we must ask the question about the proposed USP <1058>: Is it any better? To answer this question, let’s return to the four problems with the current version that I outlined earlier in this column and see.
Problem 1: Users Are Responsible for DQ
This problem has been corrected in the proposed update. In the responsibilities section, there is the statement that users are ultimately responsible for instrument qualification and validation. Users can subcontract work internally or externally, but they cannot escape overall responsibility and accountability for work carried out on their behalf.
Problem 2: The True Role of the Manufacturer Is Missing
This is improved in the new version of the general chapter. First, manufacturer is expanded to include supplier, service agents, and consultants to reflect what happens in real life. Second, manufacturers have to develop meaningful specifications for instruments. We will return to instrument specifications in the next section.
Problem 3: Poor Software Validation Guidance
This is much improved. Gone is the section on standalone software and the reference to the FDA guidance on General Principles of Software Validation (22). In its place is a tighter focus on the software used in group B and C instruments. The realization that firmware in Group B instruments can also perform calculations that need to be verified to 211.68(b) (25) or can allow users to define programs that need to be specified, controlled, and verified. There is the inclusion of more definition of the software that controls, acquires, and processes data with Group C instruments. Furthermore, the proposed guidance also references the more appropriate GAMP 5 and the associated Good Practice Guide second edition for laboratory computerized systems (20,23) for further information.
Problem 4: Groups Can Be Misleading
The approach in the proposed update to <1058> (18) is to focus on the definitions of the groups. The text makes the point that the same item can be in one or more of the groups depending on the intended use and therefore a risk assessment is essential to understand the group an item should be placed in. One such risk assessment was published in this column (17).
As you can see, these problems with the current version have been addressed by the proposed revision. Four ticks in four boxes! Therefore, is all well with the world, right? Not quite. The problem comes from a disgusting four-letter word-please sit down as this may give you palpitations. That word is user. More specifically user and his or her interaction with the 4Qs model during the DQ stage, as we shall now discuss.
A Different View of the 4Qs Model
Typically, the instrument qualification life cycle is depicted as a linear model, especially in USP <1058> (1) where it is presented in a table. However, to fully understand the 4Qs model it is better if the first three phases are presented in the form of a V as shown in Figure 1 (24).


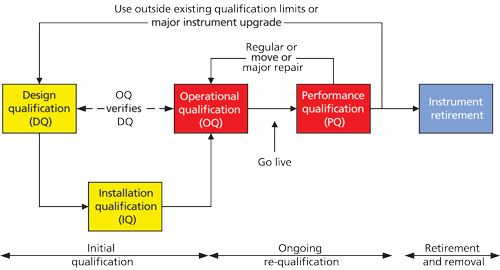
The reason is that I am often asked why analytical chemists need to write a specification for a commercial instrument. There is the supplier instrument specification that you can use. Some users think this is being smart and will save time and is all you need to do for DQ. However, in many cases, smart is being stupid. To understand why, let’s look at Figure 1 in more detail. In this simplified V model, it is much easier to see the relationship between the stages of the 4Qs as it shows the critical relationship of the DQ to the OQ: specification of intended use versus demonstration of intended use. Why do I need to write a specification? The answer is very precise and simple-it’s the law. Read the regulation you work to, either good manufacturing practice (GMP) or good laboratory practice (GLP), and it requires that equipment (25,26) or apparatus (27) to be fit for its intended use. The proposed changes to <1058> note that for commercial, off the shelf instruments, it is expected that DQ requirements would be minimal (18). This must not be interpreted as nothing. Also, it should also not always be interpreted as using the supplier’s specification unless you know how the specification was measured: will these conditions apply in your situation? The problem is that DQ is usually either not done at all or it is done badly. The OQ verifies the specification as outlined in the definition of OQ in USP <1058> (1), provided that whoever performs the OQ knows the content of the DQ or that the DQ has actually been written. There is also an ongoing dynamic requirement to manage the DQ that is reflected in the proposed <1058> (18). In part, this is dependent on how the DQ is defined (for example, if this references a very specific pharmacopeia requirement or chapter then each time the pharmacopeia is updated, the DQ for the instrument will need to be reviewed). Instead, it is more efficient to address any high-level compliance in the procedures that support the DQ and to limit the DQ requirements to actual instrument usage (24). This diagram also highlights that where an instrument has a major upgrade (as there is a wide divergence of opinion this is left to an individual laboratory to define) or is used with new methods not previously considered, there is a need to review the DQ for suitability. Without this feedback loop, there is a risk that the instrument is not suitable for a new application. Additionally, because of the relationship between the DQ and OQ, it will also highlight if different set points need to be included in the OQ to test the range of use. In terms of <1058> and risk, the OQ can cover (24) the following points:
- Qualification of a new instrument
- Requalification of an existing instrument following a defined time period typically linked with a preventative maintenance service
- Requalification following a major repair
- Extending the operating range of a qualified instrument because of an upgrade or new application that operates outside of the existing range
- Significant move of a qualified instrument with the justification for the extent of OQ testing documented in a risk assessment
The OQ is intended to demonstrate that at a fixed time point the instrument operates to the specifications in the DQ to demonstrate that it meets the intended purpose (24). At this point, it is worth repeating the statement from USP <1058> that “routine analytical tests do not constitute OQ testing” (1).
Summary
This column has discussed the changes in the proposed update to USP <1058>. The update addresses the main areas of concern with the existing version of the general chapter and this should be welcomed by the pharmaceutical industry. Table I shows the main changes between the two versions, which should not be used as the only understanding of the changes that have been proposed. In addition, we discussed the main problem with the 4Qs model-writing the design qualification document to specify the intended use of the instrument or system. This is an area for improvement throughout the industry. You have never purchased an instrument that did not meet your expectations, have you?
References
(1) General Chapter <1058> “Analytical Instrument Qualification” in United States Pharmacopeia 37–National Formulary 32 (United States Pharmacopeial Convention, Rockville, MD, 2014).
(2) “Analytical Instrument Qualification,” AAPS white paper, 2004.
(3) M. Freeman, M. Lang, D. Morrison, and R.P. Munden, Pharm. Technol. Eur.10(11), 45–48 (1995).
(4) C. Burgess, D.G. Jones, and R.D. McDowall, Analyst 123, 1879–1886 (1998).
(5) P.Bedson and M. Sargent, J. Accreditation and Quality Assurance1, 265–274 (1996).
(6) Guidance on Equipment Qualification of Analytical Instruments: High Performance Liquid Chromatography (HPLC), June 1998, LGC/VAM/1998/026.2.
(7) Guidance on Equipment Qualification of Analytical Instruments: UV-Visible Spectro(photo)meters (UV-Vis) Version 1.0 – September 2000, LGC/VAM/2000/079.
(8) R.D. McDowall, Spectroscopy25(11), 24–29 (2010).
(9) R.D. McDowall, Spectroscopy25(9), 22–31 (2010).
(10) R.D. McDowall, Spectroscopy24(12), 67–72 (2009).
(11) R.D. McDowall, Spectroscopy24(4), 20–27 (2009).
(12) R.D. McDowall, Spectroscopy21(12), 90–110 (2006).
(13) R.D. McDowall, Spectroscopy21(11), 18–23 (2006).
(14) R.D. McDowall, Spectroscopy21(4), 14–31 (2006).
(15) Curate’s egg: http://www.en.wikipedia.org/wiki/Curate’s_egg/.
(16) C. Burgess and R.D. McDowall, Pharmacopoeial Forum39(1), January-February 2012.
(17) C. Burgess and R.D. McDowall, Spectroscopy28(11), 20–27 (2013).
(18) General Chapter <1058> “Analytical Instrument Qualification” in process revision, Pharmacopeial Forum41(3), May–June 2015.
(19) ISPE, Good Automated Manufacturing Practice (GAMP) Guide version 4, (International Society of Pharmaceutical Engineering, Tampa, Florida 2001).
(20) ISPE, Good Automated Manufacturing Practice (GAMP) Guide, version 5 (International Society of Pharmaceutical Engineering, Tampa, Florida, 2008).
(21) L. Vuolo-Schuessler, M.E. Newton, P. Smith, C. Burgess, and R.D. McDowall, Pharm Eng.34(1), 46–56 (2014).
(22) US Food and Drug Administration, Guidance for Industry, General Principles of Software Validation (FDA, Rockville, Maryland, 2002).
(23) ISPE, GAMP Good Practice Guide Validation of Laboratory Computerized Systems, 1st Edition (International Society of Pharmaceutical Engineering, Tampa, Florida, 2005).
(24) P. Smith and R.D. McDowall, LCGC Europe,28(2), 110–117 (2015).
(25) Current Good Manufacturing Practice Regulations for Finished Pharmaceutical Goods, in 21 CFR 211 (U.S. Government Printing Office, Washington, DC, 2008).
(26) Good Laboratory Practice for Non-Clinical Studies, in 21 CFR 58 (U.S. Government Printing Office, Washington, DC, 2008).
(27) Principles of Good Laboratory Practice, Organisation for Economic Cooperation and Development, Paris, 1998.



R.D. McDowall is the Principal of McDowall Consulting and the director of R.D. McDowall Limited, as well as the editor of the “Questions of Quality” column for LCGC Europe, Spectroscopy’s sister magazine. Direct correspondence to: spectroscopyedit@advanstar.com












