Combining Novel and Traditional Ionization Methods for Mass Spectrometry for More Comprehensive Analyses
Special Issues
Novel ionization processes provide gas-phase ions of a wide variety of materials using MS. These simple and sensitive methods operate from solution or a solid matrix. Both manual and automated platforms are described that allow rapid switching between the ionization methods of MAI, SAI, vSAI, and conventional ESI.
Novel ionization processes provide gas-phase ions of a wide variety of materials using mass spectrometry (MS). In the case of inlet ionization, the sub-atmospheric pressure region of the mass spectrometer becomes the "ion source." These simple and sensitive methods operate from solution or a solid matrix. Matrix-assisted ionization mass spectrometry (MAI-MS) uses a solid matrix spontaneously producing gas-phase analyte ions upon exposure to sub-atmospheric pressure without the need for any external energy input. The solvent-based method of solvent-assisted ionization mass spectrometry (SAI-MS) ionizes certain classes of compounds more efficiently than electrospray ionization mass spectrometry (ESI-MS), and voltage solvent-assisted ionization mass spectrometry (vSAI-MS) is a highly sensitive combination of ESI and SAI. Direct analysis of tissue, fluids, including whole blood and urine, biological extracts, reaction mixtures, and buffered, salty solutions detecting drugs, metabolites, lipids, and proteins, simultaneously, demonstrates the advantages of the multi-ionization concept for obtaining more comprehensive analyses from 384-well plates and, if desired, hyphenated with liquid chromatography (LC), further simplifying complexity. The methodology developed for inlet ionization allows rapid switching between MAI, SAI, vSAI, and conventional ESI increasing the comprehensiveness of mass measurements at equal or better mass and drift time resolution.
Advancing chemical analysis through fundamental research and applying this knowledge to improve analyses in biological sciences will impact areas such as medical diagnostics (1). Mass spectrometry (MS), because of its specificity, sensitivity, dynamic range, wide applicability, and potential for high-throughput, is widely used in biological analyses. MS directly provides atomic and molecular composition information of heterogeneous bulk samples and surfaces, important for educated decisions on materials composition associated with quality, performance, safety, and more recently disease diagnostics. Integral to advances in MS are developments for transferring molecules into the gas phase regardless of size or volatility. As was demonstrated with electrospray ionization (ESI) and matrix-assisted laser desorption–ionization (MALDI), successful new ionization methods can have an impact on science far beyond anything envisioned in their early discovery, as witnessed by the award of a portion of the Nobel Prize in Chemistry to John Fenn and Koichi Tanaka in 2002. ESI and MALDI have strengths and weaknesses relative to analytical needs, which is the case with all ionization methods used with MS. Mass spectrometer manufacturers have recognized the need for multiple ionization methods by designing ion sources capable of ESI, atmospheric pressure chemical ionization (APCI), and atmospheric pressure photoionization (APPI) as well as offering AP-MALDI and intermediate pressure MALDI on some of these instruments. Ambient ionization methods such as atmospheric solids analysis probe (ASAP) (2), desorption electrospray ionization (DESI) (3), and direct analysis in real time (DART) (4) have also become popular. In this article, we describe new ionization techniques and manual and automated approaches that allow rapid switching between these ionization techniques as well as with ESI. The combination of methods allows interfacing with liquid separation methods, as well as direct analysis of samples, and increases the likelihood that important compounds will not be missed because of ion suppression or poor ionization efficiency.
New means of transferring small, large, volatile, and nonvolatile compounds from the solid or liquid state directly into gas-phase ions were discovered through fundamental research and developed into methods providing exceptional sensitivity and simplicity (5). The initial discovery began using laser ablation of common MALDI matrices at AP, but instead of singly charged ions expected of MALDI, multiply charged ions expected of ESI were observed (6,7). Because the mass spectra looked like ESI but used laser ablation, the method was termed laserspray ionization (LSI) (Figure 1a). Fundamental studies led to the unanticipated finding that the laser is unnecessary because analyte ions are formed when neutral matrix–analyte enters a heated inlet tube of a mass spectrometer (Figure 1b) (8,9). Further studies showed that the matrix could be the solvent used to dissolve the analyte (Figure 1c). Use of a solid matrix was termed matrix-assisted ionization (MAI) (10) and use of a solution was called solvent-assisted ionization (SAI) (11,12). While the method of introduction of the matrix sample into the heated inlet tube, which becomes the ion source, differs in LSI, MAI, and SAI, the results relative to charge states are the same, and thus the representative term inlet ionization (13,14). However, the heated inlet tube was found not to be necessary provided that heat is applied through laser absorption by the matrix, or astonishingly, without any added external energy by using an appropriate volatile matrix (15,16). In all cases, multiply protonated analyte ions are formed from compounds having multiple basic sites, such as peptides and proteins, just as in ESI. Voltages, obstructions, or additives can improve ionization in these new ionization processes (9,16–21).


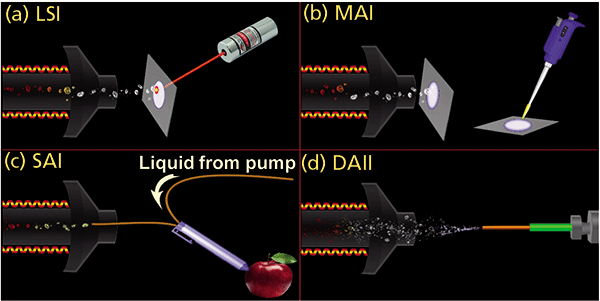
Figure 1: Graphical representations of inlet ionization methods: (a) LSI in transmission geometry, (b) MAI vacuum cleaner, (c) SAI liquid junction pen, and (d) DAII (droplet assisted inlet ionization).
Experimental
Materials
Drugs (verapamil, fexofenadine, erythromycin, azithromycin, and hydroxychloroquine), peptides (leucine-enkephalin, bradykinin, and angiotensin I), proteins (bovine insulin, ubiquitin, and cytochrome c), LB broth "Lennox", and 3-nitrobenzonitrile (3-NBN) were purchased from Sigma Aldrich. High performance liquid chromatography-grade water, methanol, acetonitrile, and dimethyl sulfoxide (DMSO) were purchased from Fisher Scientific Inc. and absolute ethanol from Decon laboratories. All chemicals were used without further purification.
Sample Preparation
Stock solutions of drugs were prepared in ethanol (verapamil and azithromycin), 1:1 acetonitrile–water (erythromycin and hydroxychloroquine), and DMSO (fexofenadine). For peptides and small proteins, stock solutions of 1 mg/mL were prepared in water. Dilutions were made in 50:50 methanol–water (drugs) and in water (peptides and small proteins) to a final concentration of 5 µM, unless otherwise noted in the text. For MAI using the automation platform, protein samples were prepared in 1:1 (v:v) methanol–water solutions and 100 mg of 3-NBN matrix was prepared in 3 mL of 3:1 (v:v) acetonitrile–water solution. For the bacteria extraction protocol, 50 mL of bacterial strains was grown to stationary phase from overnight starter cultures at 37 °C and 200 rpm on LB media. The cultures were pelleted at 4 °C for 15 min at 4000× g and washed 3× times with cold water. The pellets were resuspended in double distilled (dd) water and lysed with an ultra-sonicator by alternating 10 s on/off cycle while on ice. Cell debris was separated by centrifugation at 16,000g, 4 °C for 15 min and 1.0 mL of the supernatant collected with two methanol–water extractions and dried on a biodryer, frozen, and lyophilized. The samples were then resuspended in ethanol. For the MALDI source, the matrix–analyte mixture was prepared in 1:3 (v:v) and 1 µL of the mixture was spotted on the sample plate.
Instrumentation and Data Acquisition
Thermo Fisher Scientific Orbitrap Exactive and Waters Synapt G2S mass spectrometers were used. The Orbitrap Exactive was operated at 100,000 mass resolution (mass-to-charge [m/z] 200, 50%). The Synapt G2S instrument was operated in mobility-time-of-flight (TOF), sensitivity, and positive-ion mode detection. MAI-MS using the MSTM multifunctional automation platform was operated on the Synapt G2S mass spectrometer. The MSTM automation platform replaced the Waters ion source housing and was mounted on the instrument using a specially designed flange (22). For the automation measurements of proteins, 0.2 µL of 3-NBN and 0.1 µL of the protein solution was aspirated using a fused-silica capillary (internal diameter [i.d.] ~100 µm), and upon dispensing at the tip of the fused silica capillary allowed to dry for a few seconds prior to injection into a modified inlet tube for mass spectral analysis. For the bacterial strain, mass spectra were obtained using ESI by spraying 1.0 µL of the extract with 3 kV applied on the source, and MAI using 0.1 µL of the extract combined with 0.1 µL of the 3-NBN matrix solution, air dried and then transferred into the modified inlet. The source temperature was set at 80 °C. For MAI-MS using the MALDI source, no laser and low energy settings were used: sample plate 0–10 V, extraction 10 V, hexapole 10 V, and aperture "0" 5 V.
Results and Discussion
For those mass spectrometers that provide a heated inlet tube, ionization is readily achieved for any of the inlet ionization methods after the source is removed for access to the inlet aperture of the mass spectrometer. Examples are shown in Figure 1, with and without the use of a laser, in which various surfaces were sampled using, for example, a pen through which fused-silica tubing was held in place to form a liquid junction (Figure 1c). The solution, after contacting a surface, was sucked into the section of fused silica tubing, the exit end of which was inside the heated inlet tube and, therefore, at sub-AP (22) similar to previous studies (23,24).
Some of the small-molecule MAI matrix compounds, especially those which spontaneously produce analyte ions when exposed to sub-AP, have no labile hydrogen atoms and thus cannot directly donate protons (25). A common feature of all matrices which spontaneously produce analyte ions when subjected to sub-AP is that they sublime under the conditions of the experiment (26). Without sublimation, no ions are observed from these matrices. It is therefore surprising that such a fundamental chemical process as sublimation might still harbor secrets. Protic solvents, such as water or methanol, present in at least a few percent, enhance ionization in MAI, and especially of a nonvolatile analyte. However, drying the matrix sample prior to insertion into sub-AP does not hinder the ionization process, thereby demonstrating that ionization does not occur through liquid droplets. Studies to date suggest solid matrix particles are the engine of charge separation and the solvent the proton source. This means that ESI-like charge states are produced not only from charged liquid droplets as in ESI, but also from solid charged particles. On the other hand, SAI, in which the matrix is the solvent used to dissolve the analyte, produces ions most effectively in a heated inlet tube linking AP and the vacuum of the mass analyzer without application of a voltage, and must produce ions through highly charged liquid droplets similar to ESI. However, SAI and ESI ionize many compounds with different efficiencies. A hybrid method that combines the attributes of ESI and SAI, and termed voltage SAI (vSAI), where a voltage is applied to a solution introduced directly into a heated inlet tube through fused-silica tubing, enhances sensitivity and combines attributes of ESI and SAI. The potential for high-throughput and automated analyses were previously demonstrated for MAI and SAI (27,28), and tissue imaging applications for LSI (29–31).
The inlet ionization methods of MAI, SAI, and vSAI offer highly sensitive direct ionization methods using solid matrices like MALDI, but without a laser, a solution like ESI, but without the need of a voltage, and the solution method with a voltage that combines attributes of ESI and SAI. Each of these ionization methods offers compound-specific ionization efficiencies. For example, SAI has been shown to ionize certain steroids more efficiently than ESI (32) and MAI has low ionization efficiency for compounds that typically ionize by metal cation attachment and high efficiency for ionization of compound classes such as drugs and peptides. MAI, using 3-NBN as a matrix, ionizes typical background compounds with a low efficiency, which can be advantageous for trace analyses as was demonstrated for a sample of the peptide angiotensin I consuming just 5 femtomoles to produce a clean mass spectrum (33). This attribute makes MAI ideal for the analysis of urine, blood, and tissue samples for drugs and their metabolites, and proteins from buffered and detergent conditions, as demonstrated for the Ebola virus protein and bacteriorhodopsin, a membrane protein, as well as others (34–37). Metal cation adducts or other undesired attachments, for example matrix molecules, produced using MALDI or ESI are not observed with MAI matrices. Negative ions are spontaneously produced allowing, for example, detection of ganglioside and other lipids, such as cardiolipins, directly from a simple mitochondria extract, and carbohydrate conjugates as singly and multiply deprotonated ions with minimal fragmentation, which is often not the case using ESI or MALDI (38,39). Combining these new ionization methods with ESI in a single, easy-to-use platform gives the user the best chance to rapidly analyze compounds easily missed if only ESI, only MALDI, or only a single inlet ionization method are used for the analysis.


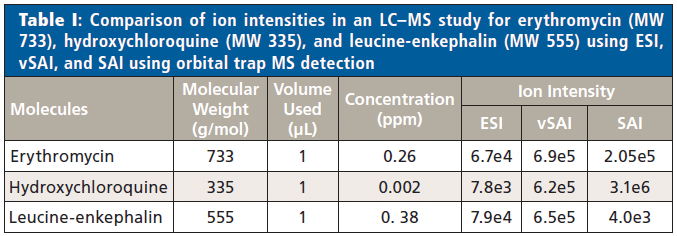
With the manual platform, the liquid introduction methods of ESI, SAI, and vSAI can be interfaced with an liquid chromatography (LC) system for LC–MS analyses. Ion intensities, in an LC–MS study, using each of the liquid introduction methods with the drugs, erythromycin (MW 733) and hydroxychloroquine (MW 335), and the peptide, leucine-enkephalin (MW 555), were determined for injection of 1 µL of solution (Table I). The same ion intensity trends were observed at higher concentrations, vividly showing differences in ionization efficiencies between the ionization methods. Additional supporting information relative to ionization efficiencies of ESI, SAI, and vSAI using LC–MS has been provided in a previous publication (19).


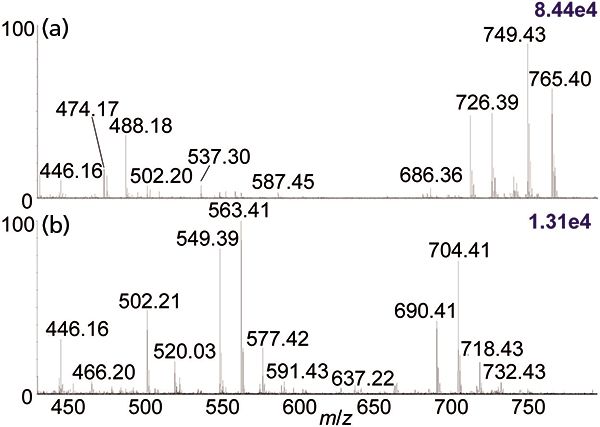
Figure 2: Comparison of MV1184 E. coli bacterial extract in 100% ethanol using the automated platform (22) and ionized by (a) ESI-MS and (b) MAI-MS on a Synapt G2S instrument.
As might be expected, there are also differences in mass spectra obtained by direct ionization using MAI, MALDI, and ESI. The differences are best seen when comparing complex mixtures. One example is the comparison of ESI and MAI using the automated system which is capable of analyzing 384-well microtiter plates or surfaces, for example glass plates, using ESI, MAI, SAI, or vSAI, just as with the manual system. Three strains of E. coli bacteria were grown, lysed, extracted in ethanol, and five replicates of each extract were acquired by MS using MAI, with the 3-NBN matrix in 3:1 acetonitrile–water, as well as with ESI directly from the ethanol extract. The results for the m/z range 400–800 of MV1184 E. coli in ethanol are shown in Figure 2. Some of the differences observed are ionization by Na+ adduction in ESI versus protonation in MAI, for example m/z 726 and 704, while other ions, for example m/z 749 and 765 in ESI and ions around m/z 637 in MAI, are not observed in both mass spectra. Because protonated ions fragment more readily, the MAI mass spectrum shows mass differences of m/z 141 (m/z 690 to 549 and m/z 704 to 563), likely representing loss of the polar head group from a glycerophospholipid. Thus, some of the contents of this extract as viewed by these two ionization methods would seem to be different.


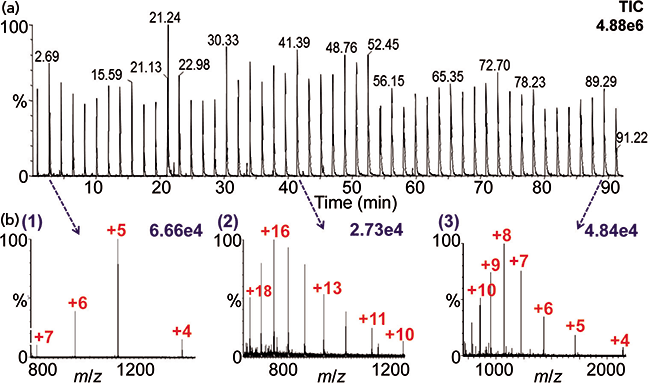
Figure 3: (a) Total ion chromatogram of 50 acquisitions and (b) MAI mass spectra of protein solutions using 3-NBN: (1) bovine insulin (MW 5733), (2) cytochrome c (MW 12384), and (3) ubiquitin (MW 8560). The mass spectra were acquired using the automated platform (22) on a Synapt G2S instrument.
An example of an automated acquisition of three proteins multiple times is shown in Figure 3 using MAI with 3-NBN as a matrix. Note that the mass spectra look nearly identical in charge states to ESI, but here ionization is from a solid rather than a liquid matrix. MAI has some distinct advantages relative to ESI. For example, simply using a different matrix provides different selectivity allowing ionization to be tailored to the problem, similar to MALDI. The most studied MAI matrices of the over 40 discovered so far are 3-NBN, 1,2-dicyanobenzene, 2-bromo-2-hydroxy-1,3-propandiol, and 2-methyl-2-hydroxy-1,3-propandiol, two of which have no labile hydrogen atoms, and yet all matrices produce singly or multiply protonated analyte ions similar to ESI. MAI, as noted earlier, does not require a heated inlet tube if the proper matrix is used. In fact, an intermediate pressure MALDI source can be used for analysis of multiple samples without the need for a laser (40,41). Figure 4 shows the analyses of six samples within 50 s in one row of a MALDI plate with samples spaced every other well. This approach has considerable potential if the time to introduce the plate into the ionization region can be greatly reduced from the current ca. 2 min. Interestingly, MAI has been reported to have equal or better mass and ion mobility resolution than ESI and MALDI on the same and other mass spectrometers (5,27).


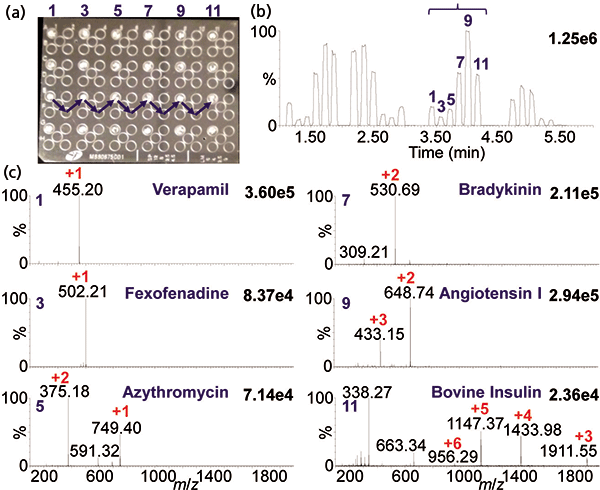
Figure 4: MAI fast analysis of six sample spots in 50 s: (a) Photograph of the sample plate with six spots (on each row) of the matrix–analyte mixture using 3-NBN as matrix, (b) total ion chromatogram, and (c) representative MAI mass spectra of the analytes from drugs (verapamil, MW 454; fexofenadine, MW 501; azithromycin, MW 748), peptides (bradykinin, MW 1059; angiotensin I, MW 1296), and small protein (bovine insulin, MW 5733). Data acquired using the commercial MALDI source of a Synapt G2S instrument.
The SAI method of ionization also has advantages over conventional ionization for certain types of analyses. As with any new discoveries, the utility becomes most apparent when others begin to apply the new technology in innovative ways. For example, the Johnston group at the University of Delaware introduced droplet assisted inlet ionization (Figure 1d), which, for some compounds, provides up to four-orders of magnitude better ion sensitivity than ESI for direct aerosol analysis (42). Professor Eberlin at the University of Texas at Austin reported on an "MS pen" device for detection of cancerous tissue during surgery using solvents as the matrix in inlet ionization (43). There are a number of other examples where solvent matrices were used without application of high voltage as required with ESI, including zero volt paper spray (44–46). Imaging applications and MS/MS of multiply charged ions directly from surfaces have also been adapted by the group of Professor Li (University of Wisconsin) (47). MAI matrices have been used with ambient ionization sources with success (48,49). Professor Murray's group at Louisiana State University constructed a pulsed valve MAI device achieving 300 times more intense signal with 3-NBN than with 2,5-dihydroxyacetophenone, a common MALDI matrix (50). The width and breadth of use of the new ionization processes indicate flexibility and broadness of the initial inventions and findings.
Conclusions
Fundamental studies relative to the new ionization technologies and methods for sample introduction continue to develop. The simplicity of ionization processes that need only exposure to the vacuum of the mass spectrometer or a heated inlet tube suggests applications where costs and simplicity are most critical. Developments in automated sample introduction with ever increasing speed and high sensitivity, and without carryover between samples or instrument contamination are being developed. Areas of application are anticipated to be in the general areas of clinical, pharmaceutical, and medical diagnostics applications, because of simplicity, speed, and robustness, but also field portable mass spectrometers because of the low pumping capacity requirements of MAI. The one thing that is clear is that the research so far has reshaped how one must think about ionization in a fundamental and applied fashion.
Acknowledgment
NSF CHE-1411376 (to ST) and NSF STTR Phase II 1556043 (to CNM) are gratefully acknowledged. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author and do not necessarily reflect the views of the National Science Foundation.
References
(1) F.G. Strathmann and A.N. Hoofnagle, Am. J. Clinical Pathol. 136, 609–616 (2011).
(2) C.N. McEwen, R.G. McKay, and B.S. Larsen, Anal. Chem. 77, 7826–7831 (2005).
(3) Z. Takats, J.M. Wiseman, B. Gologan, and R.G. Cooks, Science 306, 471–473 (2004).
(4) R.B. Cody, J.A. Laramée, and H.D. Durst, Anal. Chem. 77, 2297–2302 (2005).
(5) S. Trimpin, J. Am. Soc. Mass Spectrom. 27, 4–21 (2016).
(6) S. Trimpin, E.D. Inutan, T.N. Herath, and C.N. McEwen, Mol. Cell. Proteomics 9, 362–367 (2010).
(7) C.N. McEwen, B.S. Larsen, and S. Trimpin, Anal. Chem. 82, 4998–5001 (2010).
(8) C.N. McEwen, V. Pagnotti, E.D. Inutan, and S. Trimpin, Anal. Chem. 82, 9164–9168 (2010).
(9) J. Li, E.D. Inutan, B. Wang, C.B. Lietz, D.R. Green, C.D. Manly, A.L. Richards, D.D. Marshall, S. Lingenfelter, Y. Ren, and S. Trimpin, J. Am. Soc. Mass Spectrom. 23, 1625–1643 (2012).
(10) C.N. McEwen, V.S. Pagnotti, E.D. Inutan, and S. Trimpin, Anal. Chem. 82, 9164–9168 (2010).
(11) V.S. Pagnotti, N.D. Chubatyi, and C.N. McEwen, Anal. Chem. 83, 3981–3985 (2011).
(12) V.S. Pagnotti, S. Chakrabarty, B. Wang, S. Trimpin, and C.N. McEwen, Anal. Chem. 86, 7343–7350 (2014).
(13) C.B. Lietz, A.L. Richards, Y. Ren, and S. Trimpin, Rapid Commun. Mass Spectrom. 25, 3453–3456 (2011).
(14) C.N. McEwen, V.S. Pagnotti, E.D. Inutan, and S. Trimpin, Anal. Chem. 82, 9164–9168 (2010).
(15) E.D. Inutan, B. Wang, and S. Trimpin, Anal. Chem. 83, 678–684 (2010).
(16) E.D. Inutan and S. Trimpin, Mol. Cell. Proteomics 12, 792–796 (2013).
(17) N.D. Chubatyi, T. Wang, and C.N. McEwen, Rapid Commun. Mass Spectrom. 26, 2763–2769 (2012).
(18) V.S. Pagnotti, N.D. Chubatyi, A.F. Harron, and C.N. McEwen, Anal. Chem. 83, 6828–6832 (2012).
(19) M.A. Fenner, S. Chakrabarty, B. Wang, V.S. Pagnotti, K. Hoang, S. Trimpin, and C.N. McEwen, Anal. Chem. 89, 4798–4802 (2017).
(20) Z.J. Devereaux, C.A. Reynolds, J.L. Fischer, C.D. Foley, J.L. DeLeeuw, J. Wager-Miller, S.B. Narayan, K. Mackie, and S. Trimpin, S., Anal. Chem. 88, 10831–10836 (2016).
(21) N.D. Chubatyi and C.N. McEwen, J. Am. Soc. Mass Spectrom. 26, 1649–1656 (2015).
(22) S. Trimpin, S. Rauschenbach, K. Kern, I-C. Lu, E.D. Inutan, J.L. Fischer, S. Thawoos, Z. J. Devereaux, W.-J. Zhang, W. Zhang, M. Dillenburger, H.-J. Radar, K. Mullen, K. Hoang, A. Siderenko, M. Pophristic, and C.N. McEwen, "Fundamental Studies of Inlet Ionization with Sample Introduction at Atmospheric Pressure," paper presented at the 64th ASMS Conference on Mass Spectrometry and Allied Topics, San Antonio, Texas, 2016.
(23) B. Wang, C.L. Dearring, J. Wager-Miller, K. Mackie, and S. Trimpin, Eur. J. Mass Spectrom. 21, 201–210 (2015).
(24) S. Trimpin, B. Wang, C.B. Lietz, D.D. Marshall, A.L. Richards, and E.D. Inutan, Crit. Rev. Biochem. Mol. Biol. 48, 409–429 (2013).
(25) S. Trimpin and E.D. Inutan, J. Am. Soc. Mass Spectrom. 24, 722–732 (2013).
(26) S. Trimpin, C.A. Lutomski, T.J. El-Baba, D.W. Woodall, C.D. Foley, C.D. Manly, B. Wang, C.W. Liu, B.M. Harless, R. Kumar, L.F. Imperial, and E.D. Inutan, Int. J. Mass Spectrom. 377(SI), 532–545 (2015).
(27) B. Wang and S. Trimpin, Anal. Chem. 86, 1000–1006 (2014).
(28) D.W. Woodall, B. Wang, E.D. Inutan, S.B. Narayan, and S. Trimpin, Anal. Chem. 87, 4667–4674 (2015).
(29) A.L. Richards, C.B. Lietz, J. Wager-Miller, K. Mackie, and S. Trimpin, Rapid Commun. Mass Spectrom. 25, 815–820 (2011).
(30) E.D. Inutan, J. Wager-Miller, K. Mackie, and S. Trimpin, Anal. Chem. 84, 9079–9084 (2012).
(31) A.F. Harron, K. Hoang, and C.N. McEwen, Int. J. Mass Spectrom. 352, 65–69 (2013).
(32) N.D. Chubatyi, V.S. Pagnotti, C.M. Bentzley, and C.N. McEwen, Rapid Commun. Mass Spectrom. 26, 887–892 (2012).
(33) K. Hoang, M. Pophristic, A.J. Horan, M.V. Johnston, and C.N. McEwen, J. Am. Soc. Mass Spectrom. 27, 1591–1596 (2016).
(34) E.D. Inutan, J. Wager-Miller, S.B. Narayan, K. Mackie, and S. Trimpin, Int. J. Ion Mobility Spectrom. 16, 145–159 (2013).
(35) D.D. Marshall, E.D. Inutan, B. Wang, C.W. Liu, S. Thawoos, J. Wager-Miller, K. Mackie, and S. Trimpin, Proteomics 16, 1695–1706 (2016).
(36) S. Trimpin, S. Thawoos, C.D. Foley, D.W. Woodall, J. Li, E.D. Inutan, and P.M. Stemmer, Methods 104, 63–68 (2016).
(37) S. Thawoos, J. Wager-Miller, E.D. Inutan, Z.J. Devereaux, C.D. Foley, Y.-H. Ahn, K. Mackie, P.M. Stemmer, and S. Trimpin, "Rapid Analysis of Proteins on High-Resolution Mass Spectrometers using Matrix-Assisted Ionization," paper presented at the 64th ASMS Conference on Mass Spectrometry and Allied Topics, San Antonio, Texas, 2016.
(38) C. Reynolds, J. DeLeeuw, C. Corinne Lutomski, T. Sanderson, K. Przyklenk, and S. Trimpin, "Matrix-Assisted Ionization-Mass Spectrometry Enables Cardiolipin Characterization Directly From Intact Mitochondrial Membranes," paper presented at the 63rd ASMS Conference on Mass Spectrometry and Allied Topics, St. Louis, Missouri, 2015.
(39) B. Wang, G. Liao, Z. Guo, and S. Trimpin, "Characterization of Carbohydrate-Monophosphoryl Lipid Conjugate Cancer Vaccine Candidates using Matrix Assisted Ionization Vacuum Mass Spectrometry," paper presented at the 62nd ASMS Conference on Mass Spectrometry and Allied Topics, Baltimore, Maryland, 2014.
(40) S. Trimpin and E.D. Inutan, J. Am. Soc. Mass Spectrom. 24, 722–732 (2013).
(41) E.D. Inutan and S. Trimpin, Mol. Cell. Proteomics 12, 792–796 (2013).
(42) A.J. Horan, M.J. Apsokardu, and M.V. Johnston, Anal. Chem. 89, 1059–1062 (2017).
(43) J. Zhang, J. Rector, J.Q. Lin, J.H. Young, M. Sans, N. Katta, N. Giese, W. Yu, C. Nagi, J. Suliburk, J. Liu, A. Bensussan, R.J. DeHoog, K.Y. Garza, B. Ludolph, A.G. Sorace, A. Syed, A. Zahedivash, T.E. Milner, and L.S. Eberlin, Sci. Transl. Med. 9, eaan3968 (2017).
(44) A. Motoyama and K. Kihara, Rapid Commun. Mass Spectrom. 29, 1905–1916 (2015).
(45) P. Wei, S. Bag, C.J. Pulliam, D.T. Snyder, and R.G. Cooks, Anal Methods 8, 1770–1773 (2015).
(46) M. Wleklinski, Y. Li, S. Bag, D. Sarkar, R. Narayanan, T. Pradeep, and R.G. Cooks, Anal. Chem. 87, 6786–6793 (2015).
(47) B. Chen, C.B. Lietz, and L. Li, J. Am. Soc. Mass Spectrom. 25, 2177–2180 (2014).
(48) K. Kanaki and S.A. Pergantis, Rapid Commun. Mass Spectrom. 28, 2661–2669 (2014).
(49) J.M. Santos, P.H. Vendramini, N.V. Schwab, M.H. Eberlin, and D.R. Morais, J. Mass Spectrom. 51, 53–61 (2016).
(50) B. Banstola and K.K. Murray, Analyst 142, 1672–1675 (2017).
Sarah Trimpin is with the Department of Chemistry at Wayne State University in Detroit, Michigcan, and the Cardiovascular Research Institute at Wayne State University School of Medicine. Santosh Karki, Darrell D. Marshall, Ellen. D. Inutan and Anil K. Meher are with the Department of Chemistry at Wayne State University. Sara Madarshahian, Madeline A. Fenner and Charles N. McEwen are with the Department of Chemistry & Biochemistry at the University of the Sciences in Philadelphia, Pennsylvania. Direct correspondence to: strimpin@chem.wayne.edu













LIBS Illuminates the Hidden Health Risks of Indoor Welding and Soldering
April 23rd 2025A new dual-spectroscopy approach reveals real-time pollution threats in indoor workspaces. Chinese researchers have pioneered the use of laser-induced breakdown spectroscopy (LIBS) and aerosol mass spectrometry to uncover and monitor harmful heavy metal and dust emissions from soldering and welding in real-time. These complementary tools offer a fast, accurate means to evaluate air quality threats in industrial and indoor environments—where people spend most of their time.


NIR Spectroscopy Explored as Sustainable Approach to Detecting Bovine Mastitis
April 23rd 2025A new study published in Applied Food Research demonstrates that near-infrared spectroscopy (NIRS) can effectively detect subclinical bovine mastitis in milk, offering a fast, non-invasive method to guide targeted antibiotic treatment and support sustainable dairy practices.

Smarter Sensors, Cleaner Earth Using AI and IoT for Pollution Monitoring
April 22nd 2025A global research team has detailed how smart sensors, artificial intelligence (AI), machine learning, and Internet of Things (IoT) technologies are transforming the detection and management of environmental pollutants. Their comprehensive review highlights how spectroscopy and sensor networks are now key tools in real-time pollution tracking.


