CSA: Much Ado About Nothing?
Computerized system validation (CSV) in good practice (GxP)-regulated laboratories is alleged to take ages and generate piles of paper to keep inspectors and auditors quiet. To the rescue is the draft FDA guidance on computer software assurance (CSA). But...is this really the case?
I wrote about the Food and Drug Administration (FDA)’s computer software assurance (CSA) in an earlier “Focus on Quality” column (1). I was not complimentary of the CSA, because it was “regulation” by presentation and publication, as the FDA had failed to issue the draft guidance, which I likened to waiting for Godot. Well, Godot has arrived! FDA issued the draft guidance on September 12th, with an effective date of September 13th (2). The Center for Devices and Radiological Health (CDRH) is the originator in consultation with Center for Drug Evaluation and Research (CDER), but only medical device good manufacturing practice (GMP) regulations are mentioned in the guidance.
It is good that the guidance is available so that claims of the more outspoken advocates can be verified. In this article, we explore if computerized system validation (CSV) is to be cast into the recycling bin of history with CSA ruling the roost, or whether CSA and CSV live together happily ever after, especially as the aim of both is that computerized systems are fit for their intended use. I discuss this guidance from the perspective of good practice (GXP)-regulated laboratories in the pharmaceutical industry.
My Philosophy for Validation
In the interests of transparency, and to put my comments into perspective, let me outline my philosophy for implementing and validating laboratory computerized systems in the pharmaceutical industry:
1. There is no point in implementing and validating a computerized system if you do not gain substantial business benefits, as noted by in Clause 5.6 of the 2016 World Health Organization (WHO) guidance (3). Otherwise, why would an organization bother wasting money on implementing a slow and error-prone hybrid approach? Deriving business benefits involves redesigning the business process, and using electronic signatures coupled with eliminating spreadsheet and calculator calculations and paper printouts. Interfacing the system with other informatics applications will leverage even greater process efficiencies. Yes, this takes more time, but the payback on the overall investment is immense.
2. Performed correctly, the first phase of a validation life cycle (selection) is investment protection and the second phase (implementation) is simply good software engineering practice. Documenting user requirements ensures you purchase and implement the right instrument and application (4,5). Documenting the configuration and settings provide you with the baseline for change control and ensures data integrity. It is also the ultimate backup in case of a major disaster, as you’ll be able to reinstall and reconfigure the application quickly.
3. Leverage a supplier’s software development to reduce user acceptance testing subject to an on-site or virtual assessment with a report (6). Many functions in laboratory systems are parameterized and not configured (including temperature and detector wavelength), and the business process does not change. If the supplier has developed and tested these functions adequately, why would you want to test them as noted by GAMP 5 (7,8)?
4. Manage risk effectively. GAMP risk assessment is not the only methodology you can use (9). Furthermore, nobody can test any computerized system completely—not even a supplier. Users should focus on demonstrating that the system works as intended rather than finding software errors. See the big picture for the system: will it be connected to another laboratory informatics application in the future, or remain as a standalone system? Conversely, avoid adding “just in case” user requirements that you may never use. Finally, document your assumptions, exclusions, and limitations for your user acceptance testing (10).
5. Flexibility or scalability in your validation approach is essential, as one size does not fit all. You need to tailor your validation to the risk posed by each system and the records generated by it. As regulations, citations, guidance, and technology are changing, there are always better ways to perform the next validation. Maintaining a status quo CSV approach is not an option.
This philosophy above is based on process efficiency and investment protection, while regulations are only mentioned in point 5. If you approach CSV correctly, then business efficiencies pay for the system and the validation costs.
CSA: The Promised Land
Before the release of the draft guidance, CSA was proposed as a better approach to demonstrating fitness for intended use by avoiding the generation of piles of paper to keep auditors and inspectors quiet. The aims were:
- Focus on critical thinking rather than documentation and testing formalization;
- Define the intended use of the software and determine the impact on product quality and patient safety;
- Identify any functions within the software that impact product quality, patient safety, or data integrity, and determine the risk on them;
- Leverage supplier development and documentation where possible; and
- Define assurance activities through conducting scripted and unscripted testing activities based on risk.
Now that we have the draft guidance available, we can assess how these promises stack up with reality. Note that the maturity of an organization in interpreting intended use, risk assessment, and testing will impact if they streamline their current CSV approaches or not.
Meet the New Boss...
The title of the draft guidance is “Computer Software Assurance for Production and Quality System Software (2).“
The draft guidance consists of 25 pages. We find on page 4 that CSA is not a universal panacea, a change to the regulatory paradigm, an approach that turns the world upside down, or a revolution that replaces CSV—only that CSA is a supplement to an existing 20-year-old FDA guidance. What?! Despite of all the trumpeting of how superior CSA will be, the “General Principles of Software Validation (GPSV) (11),” issued in 2002, will be the master, and CSA only a slave. When finalized, the CSA guidance (11) simply replaces three pages of Section 6 with about 20 pages (see Figure 1). Green represents the sections of the GPSV that will remain effective, red represents the three sub-sections of that will be replaced by the CSA guidance, and yellow shows how Section 6 will look when the CSA guidance is finalized. This column will not consider the examples in Appendix A, as there is no direct impact on spectroscopic systems. Five points immediately arise:
1. Far from being a total replacement for CSV, CSV and CSA will coexist. However, the current Section 6 mentions both system validation and software validation—so what term is CSA replacing? CSV is required for medical device software validation under 21 CFR 820 (12) and 21 CFR 11 (13). CSA will only be focused on lower risk production and automation applications. In February 2022, FDA proposed updating 21 CFR 820 to harmonize with ISO 13485-2016 (14). It will be interesting to see if the CSA guidance is compatible with the new regulation.
2. CSA does not cover the whole gamut of software covered by CDRH, but only the lower risk software involved in production and quality systems. However, production systems are hardly mentioned in this guidance.
3. If the CSA guidance is supposed to be a supplement to the GPSV and replace section 6, why not combine the two into a single guidance? The obvious advantage is that there would be a single guidance to read rather than two, one of which has a section that has been replaced but without notifying readers. Ozun Okutan has provided a detailed rebuttal of CSA in a blog on LinkedIn. There are existing medical device software standards that are used outside the US so why not use them (15)?
4. The focus is on software—a rather amorphous term. Does this mean application, a macro, or just “software?”
5. The FDA doesn’t make it clear whether sections 3–5 of the GPSV do or no not impact section 6, so it’s a dog’s dinner of a situation.


FIGURE 1: Replacement of Section 6 of the general principles of software validation by the CSA guidance.
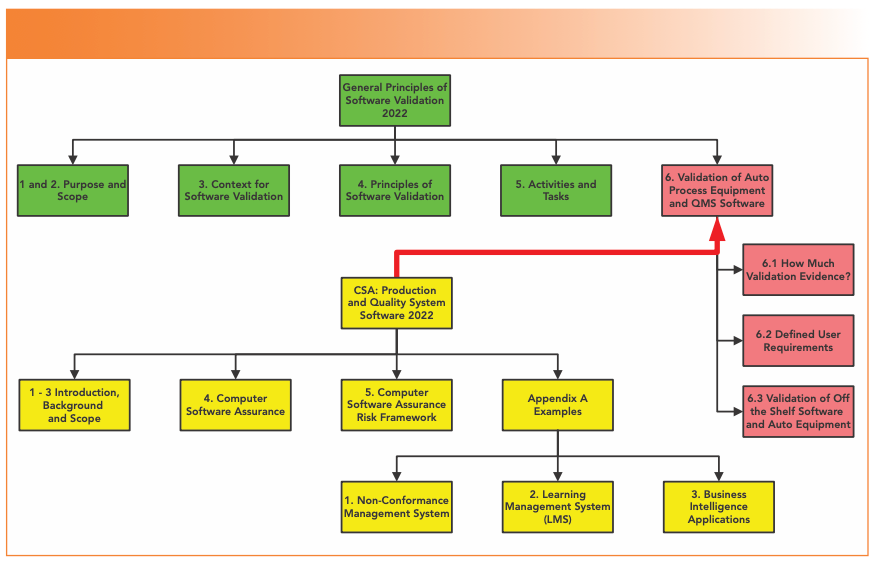
When I read the guidance for the first time, this was a shock, as some CSA advocates were talking about a paradigm shift. I was expecting a new approach; instead, the wheels appear to come off the CSA bandwagon, and the guidance is a half-baked anticlimax. Still, let’s soldier on, and see what other gems await us.
System vs. Software Validation
One confusing element of the CSA guidance is the terminology used. “Computerized system validation” includes elements of the software, computer hardware, IT infrastructure, any connected instrument, and the trained staff. Conversely, without any definition, “computer software assurance” focuses on software alone:
- Computer software assurance is a risk-based approach for establishing and maintaining confidence that software is fit for its intended use.
- Computer software assurance establishes and maintains that the software used in production or the quality system is in a state of control throughout its lifecycle (2).
Fine words, but meaningless. At least the definition of validation in the GPSV provides understanding (11).
CSA Risk Framework
The main body of the draft guidance (section V) is split into four main sections under the heading of CSA Risk Framework, shown in Figure 2.

.")
FIGURE 2: Structure of the CSA risk framework from Section V of the draft CSA Guidance (2).
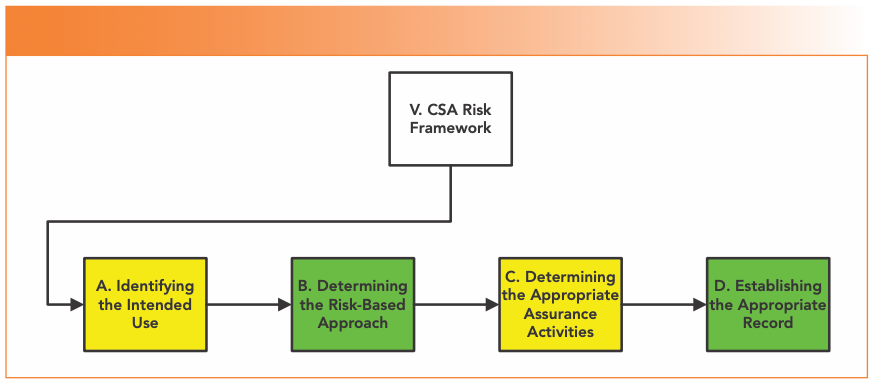
Identification of Intended Use
The first stage is to define the software’s high level intended use, which is not about individual functions within the software. This is essential, because software that is used directly or indirectly as part of production or quality system must be validated under 21 CFR 820.70(i) (12), whereas the guidance does not apply for software that does not. Direct software is stated to be:
Production Software
- Direct: Automating production processes, inspection, testing, or collection and processing of production data.
- Indirect: Development tools that test or monitor software system or automated software test tools.
Quality System Software
- Direct: Automating quality system processes, collection and processing of quality system data, or maintaining a quality record under 21 CFR 820 (12).
Software usually not requiring validation is listed as general business management and supporting infrastructure, such as networking. For the latter, there is a potential clash if applied in pharma as the Principle of EU GMP Annex 11 requires IT infrastructure to be qualified (16).
As the General Principles guidance is still effective, don’t forget the sentence in Section 5.2.2: It is not possible to validate software without predetermined and documented software requirements (11). Your user requirements specification (URS), that I interpret as “intended use,” is the means to ensure the right system for the right job and protect your investment.
As CSV uses URS, and CSA uses intended use, there is a potential for conflict and misunderstanding. It would help greatly if the CSA guidance provided advice on how to document intended use. Is definition of intended use a user requirements specification, or a replacement for it? It could also include the use of a software engineering tool to manage requirements electronically.
Intended Use or Intended Purpose?
Are these two terms interchangeable or different? It depends. If you are the United States Pharmacopoeia (USP), the two terms have different meanings. “Intended use” is applied to analytical instruments, but “intended purpose” refers to both analytical procedure and software, as they focus on the overall process. This is emerging during the revision of USP <1058> (17).
Determining the Risk-Based Approach
In the guidance, the FDA offers a binary choice here between “high process risk” and “not high process risk”—no fence sitting here—and identifies high process risk features as functions or operation automated by software. The five areas identified in the guidance are shown in Figure 3. Item 2 is the one area where laboratory systems may be involved—Process Analytical Technology (PAT), where spectroscopic systems integrated with process systems test drug content in tablets.

.")
FIGURE 3: High process risk software functions (2).
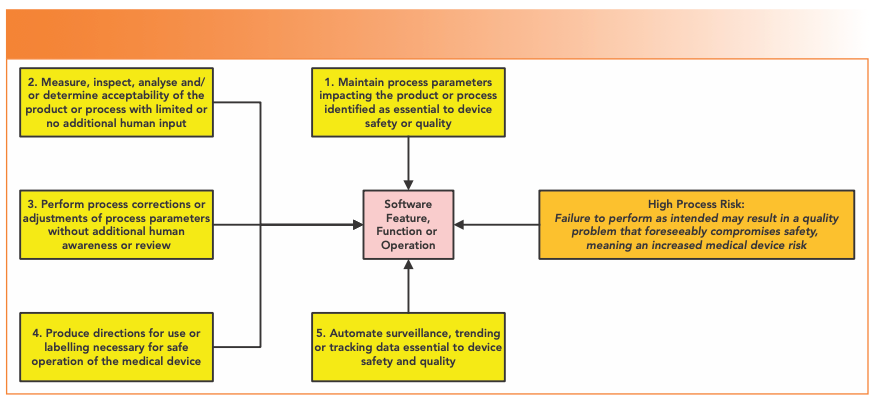
Laboratory-centric examples provided in this section to illustrate the risk-based approach are three enterprise resource planning (ERP) scenarios, a manufacturing execution system (MES), and an automated graphical user interface.
Determining the Appropriate Assurance Activities
Simply put, for high process risk functions, the higher the risk, the more validation, sorry assurance activities are required, and vice versa. Mentioned earlier in the guidance are the requirements to ensure that security, configuration settings, data storage, data transfer, and error trapping features must be set up and documented—interestingly, nothing about data integrity.
A major problem with the CSA guidance is that the word “testing” is mentioned 145 times, versus 106 times for quality. There is a potential danger of falling into the trap of trying to test quality into software rather than ensure requirements are properly defined.
One of the key CSA benefits advocated was streamlined test documentation. There are five types of assurance activity (as shown in Figure 4), which are mostly based on the general concepts of software testing in part 1 of ISO 29119-1 (18).

 in the draft CSA Guidance (2).")
FIGURE 4: The types of assurance activities (testing) in the draft CSA Guidance (2).
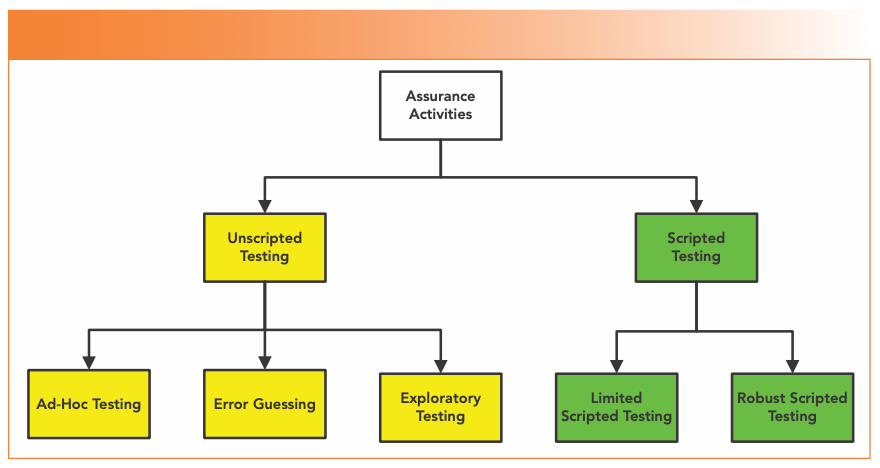
Unscripted Testing
Dynamic testing in which the tester’s actions are not prescribed by written instructions in a test case, and is outlined in ISO 29119-1 (18); it would be good if the FDA referenced the 2022 version to be current. The guidance sub-divides testing, as shown in Figure 4, into:
- Ad-hoc testing unscripted practice that does not rely on large amounts of documentation;
- Error-guessing testing based on a tester’s knowledge of past failures; and
- Exploratory testing spontaneously designs and executes tests based on the tester’s relevant knowledge and experience of the test item.
As I noted in my earlier article, prototyping is performed without documentation (1), as well as evaluating new software in a development environment.
Scripted Testing
Dynamic testing in which the tester’s actions are prescribed by written instructions in a test case (18), which are divided into:
- Limited scripted testing: A hybrid of scripted and unscripted testing scaled according to the risk passed by the system; and
- Robust scripted testing: Scripted testing that includes links to traceability to requirements and auditability
These last two scripted testing approaches are not in the ISO 29119-1 (18), but have been devised by FDA.
A problem with the guidance is extensively quoting a software and systems engineering standard ISO 29119, where testers will have a fuller picture of software and may be testing the same modules or application repeatedly or in depth. What problems can you see from the testing options above?
1. To perform error-guessing and exploratory unscripted testing, you need a knowledge of the application you are testing. If it is new system and you have just received training, how much do you know of past failures or experience of the test item? Zero? Then these two test approaches have just bitten the dust.
2. EU GMP Annex 11 clause 4.4 states, “User requirements should be traceable throughout the life cycle (16).“ Therefore, can only robust scripted testing be applied in a regulated GMP pharmaceutical laboratory? So where is the much-vaunted improvement and benefit in testing claimed by the CSA community? There appears to be none in a pharmaceutical context.
Does this mean that a coach, horses, and a safari of migrating wildebeest have been driven though a key benefit of CSA? The risk is that there is an apparent disconnect between the guidance’s approach to streamlining testing and a user’s knowledge of the application coupled with pharma GMP regulatory requirements for network qualification and requirements traceability (16). Another problem is with the ISO standard quoted for the different testing approaches.
Q: When is a Consensus Standard Not a Consensus Standard?
A: When it is ISO 29119!
FDA use some of the definitions for types of software testing from this ISO standard. Developing a consensus is important for ISO standards; however, there is a problem with ISO 29119-1 (18). The standard has been criticized as too simplistic, and does not represent a consensus for software testing (19,20). Wikipedia has a summary of the contro- versy, which has led at least two testing organizations to call for the standard to be withdrawn (21). Some of the reasons given for the withdrawal were it did not represent a consensus, it profited by selling standards, it was deemed too prescriptive, there was no need for testing standardization, and—wait for it—there was a heavy focus on documentation (21)! The draft CSA guidance appears to select some testing methodologies (and ignore others) without reason.
Establishing the Appropriate Record
In Table I of the CSA guidance, we can see the level of documentation required. It is important to note that all types of assurance activities require documentation of the work performed, but obviously the extent and detail varies. Its good to see that the old saying “If its not written down, it’s a rumor” survives at the FDA.
- Ad-hoc testing: Requires intended use and risk determination to be documented, a summary description of the testing performed, any issues found and resolved, who performed the testing and when, and review and approval (as appropriate!), plus a conclusion statement.
- Robust scripted testing: Requires intended use and risk determination to be documented, as well as the test objectives, expected results, pass/fail criteria, detailed report of testing, any issues found and resolved, who performed the testing and a conclusion statement, and also calls for review by a second person pre- and post-execution of the protocol.
An example of exploratory testing of a spreadsheet for collecting and presenting nonconformance data is discussed. A spreadsheet? You cannot be serious! Readers will know of my high regard for spreadsheets used in a regulated environment (22,23), so I’m surprised to see a spreadsheet example in an FDA guidance document. This raises serious issues of data integrity and governance, such as how to document and test requirements to show that the DI controls work. Perhaps this was a lack of critical thinking?
Completing this section is the following sentence:
As a least burdensome method, FDA recommends the use of electronic records, such as system logs, audit trails, and other data generated by the software, as opposed to paper documentation and screenshots, in establishing the record associated with the assurance activities (2).
This is best part of the whole guidance! I have no problem with this sentence, as I have used an application to self-document user acceptance testing for years (6). Why do more?
Same as the Old Boss...
In this section, I’ll look at what was promised versus what was delivered, where have we seen this before, and summarize some of the inconsistencies that we’ve seen in the guidance.
Missing in Action?
Now let us compare the CSA approach promoted in presentations and publications before in the draft guidance was issued, as shown in Table I.
Let us look at the missing approaches in more detail:
- Critical Thinking: Proposed as a major component that was lacking in CSV. Is it mentioned directly in the CSA guidance? No. Missing in action.
- Unscripted Testing: Even unscripted testing requires documentation as noted in the draft guidance
However, the impression given in some CSA presentations was that unscripted testing equated to undocumented testing.
- Leveraging Trusted Suppliers: proposed as a way of reducing testing.
Is it mentioned directly in the CSA guidance? No. Missing in action.
Leveraging suppliers is covered in ASTM E2500 (24), GAMP 5, and GAMP 5’s 2nd edition. (7,8). GAMP discusses leveraging supplier involvement in sections 2.1.5, 7 and 8.3, plus appendix M2 for supplier assessments, as noted in my earlier publication on CSA (1). 21 CFR 11.10(a) on validation could also imply using suppliers for accuracy and reliability of electronic records (13).
Why supplier involvement was omitted in the guidance is probably due to the emphasis on ISO 29119-1. The main emphasis of the FDA draft guidance appears to be on developing and testing software, and not on effective implementation of purchased applications.
- Production systems: Nothing
- Laboratory systems: Nothing
- Automation: Fine, in principle, but it takes time to learn and use the automation tool, as well as validate and assure the software. Expect the first system to take longer to validate. As many lab systems are standalone how will the automation tool cope with this?
It’s Déjà vu All Over Again!
Reading the draft guidance, why do I get the feeling that I have read this before? Let’s have a look in more detail:
1. Risk-Based Approach: The draft guidance explains that CSA is a risk- based approach, and follows a least burdensome approach.
Have we seen this before? Yes, it is in the GPSW (11) and Clause 1 of Annex 11 (16). Back to the future!
2. Define Intended Use: This has been a requirement in 21 CFR 211.63 since 1978 (25), and in 21 CFR 820.30 and 70(i) since 1996 (12). Moreover, the GPSV states in section 5.2.2 Requirements “It is not possible to validate software without predetermined and documented software requirements (11).”
In the quotation about self-documenting testing above, the proposal for using an audit trail to document test data has been in GAMP 5 since 2008. Section 8.5.3 notes:
Systems may have audit trails that capture much of the information that is traditionally captured by screen prints. If such an audit trail exists, is secure, and is available for review by the SME, then capturing additional evidence may not be necessary [7].
Same old, same old.
Regulatory Contradictions
Although CDER was consulted, there appears to be at least two contradictions adapting CSA to meet current global pharmaceutical GMP regulations:
1. Infrastructure Qualification: The principle of EU GMP Annex 11 states IT infrastructure shall be qualified (16). This is interpreted to include the software elements of infrastructure and is described in the GAMP GPG on Infrastructure Control and Compliance (26). Furthermore, backup must be validated under Annex 11 clause 7.2 (16). In contrast, the CSA guidance notes that for software intended for establishing or supporting infrastructure not specific to production or the quality system, such as networking or continuity of operations (2), the requirement for validation under 21 CFR 820.70(i) would not apply. Clarification is urgently needed here.
2. Requirements Traceability: According to the draft CSA guidance, the only robust scripted testing requires traceability of requirements (2). This is in contradiction with Clause 4.4 of Annex 11 that requires requirements traceability throughout the life cycle (16).
Don’t you just love it when regulatory authorities don’t agree?
Won’t Get Fooled Again
To summarize, while the guidance emphasizes the importance of applying software assurance during software development, my view is unchanged from my earlier publication (1): CSA is not needed. Given the lack of anything new you wonder what the FDA – Industry group was doing for six years. There is nothing for a laboratory in the CSA guidance (2), and there are contradictions with EU GMP Annex 11 (2). Additionally, organizations with a mature approach to software development and testing do not need the guidance, but there is scope for confusion and misapplying the guidance by organizations with a poor understanding of CSV principles.
Current regulations (12,27) and regulatory guidance (including least burdensome approach (11) and industry guidance (8)) provide sufficient information for an effective risk-based approach to CSV and to leverage supplier development. The problem is that the pharmaceutical industry is risk averse, and reluctant to change.
However, CSA has opened a debate on how to manage risk and how to document testing, which is good. The problem with the way that CSA has been promoted has been polarizing, and that people who disagreed were classified as contemptible. However, the CSA more has become much less with the publication of the draft guidance.
Same Old, Same Old?
Regardless of the outcome of the debate of CSV versus CSA, will the pharmaceutical industry change? To illustrate this, I would like to turn back the clock 20 years to the publication of the FDA’s GPSV. In section 3.1.3 is the following quote:
While installation qualification (IQ)/operational qualification (OQ)/performance qualification (PQ) terminology has served its purpose well and is one of many legitimate ways to organize software validation tasks at the user site, this terminology may not be well understood among many software professionals, and it is not used elsewhere in this document (1).
Furthermore, with the publication of GAMP 5 in 2008, the use of IQ, OQ, and PQ, used since the first version of the GAMP Guide, was discontinued in favor of verification (7).
But the 4Qs model is deeply embedded in the mindset of pharma manufacturers, regulators and QA. USP <1058> (4), as well as EU GMP Annex 15 (5), uses the 4Qs model, and it is a mandatory requirement for any supplier to provide IQ/OQ documents to any pharmaceutical company. The newly published stimulus article for revision of USP <1058> is an attempt in embedding the 4Qs model into an overall qualification lifecycle model (28).
Will pharma change? Probably not.
Summary
The debate about CSV and CSA is about the same subject: does a computerized system meet intended use requirements? The current draft guidance is simply rearranging the deckchairs on the Titanic. Better would be to reissue an updated version of the general principles of software validation with integrated CSA, as it is currently aimed specifically at medical devices and appears more focused on software development than system validation. Furthermore, the Agency needs to make it clear whether the CSA is just for medical devices, or applicable to pharma as well. Only a little of the current guidance can be transferred to GXP in the pharmaceutical industry. If regulated organizations and individuals had read and followed regulations and guidance published over the past 20 years, there would no need to reinvent CSV as CSA.
There is nothing in the CSA guidance about improving processes (merely auto- mating them), selecting the right application (selection is only mentioned in terms of testing) and data integrity (only features in the Appendix examples). The great CSV expert William Shakespeare summed up CSA best...much ado about nothing.
Acknowledgments
I would like to thank, in alphabetical order, Akash Arya, Chris Burgess, Markus Dathe, Orlando Lopez, Mahboubeh Lotfinia, Bunpei Matoba, Ozan Okutan, Yves Samson, and Paul Smith for their critique and comments in the preparation of this column. I appreciate all your inputs, but integrating them was something else!
References
(1) McDowall, R. D. Does CSA Mean “Complete Stupidity Assured?” Spectroscopy 2021, 36 (9), 15–22. DOI: 10.56530/spectroscopy.vl2478y4
(2) FDA Draft Guidance for Industry, Computer Software Assurance for Production and Quality System Software; Food and Drug Administration: Silver Spring, MD, 2022. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/computer-software-assurance-production-and-quality-system-software (accessed 2022-10-10).
(3) WHO Technical Report Series No. 996 - Annex 5: Guidance on Good Data and Records Management Practices; World Health Organization: Geneva, 2016. https://www.who.int/publications/m/item/trs-966---annex-5-who-good-data-and-record-management-practices (accessed 2022-10-10).
(4) USP General Chapter <1058> Analytical Instrument Qualification. United States Pharmacopoeia Convention Inc.: Rockville, MD. DOI: 10.31003/USPNF_M1124_01_01
(5) EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Annex 15: Qualification and Validation; European Commission: Brussels, 2015. https://health.ec.europa.eu/system/files/2016-11/2015-10_annex15_0.pdf (accessed 2022-10-10).
(6) McDowall, R. D. Validation of Chromatography Data Systems: Ensuring Data Integrity, Meeting Business and Regulatory Requirements, Edition 2; Royal Society of Chemistry: Cambridge, 2017. DOI: 10.1039/9781782624073
(7) ISPE. Good Automated Manufacturing Practice (GAMP) Guide Version 5; International Society for Pharmaceutical Engineering: Tampa, FL, 2008.
(8) ISPE. Good Automated Manufacturing Practice (GAMP) Guide 5, 2nd ed.; International Society of Pharmaceutical Engineering: Tampa, FL, 2022.
(9) McDowall, R. D. Effective and Practical Risk Management Options for Computerised System Validation. Qual. Assur. J. 2005, 9, 196–227. DOI: 10.1002/qaj.339
(10) IEEE Software Engineering Standard 829 - 2008 Software Test Documentation; Institute of Electronic and Electrical Engineers: Piscataway, NJ, 2008. DOI: 10.1109/IEEESTD.2008.4578383
(11) FDA Guidance for Industry: General Principles of Software Validation; Food and Drug Administration: Rockville, MD, 2002. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-principles-software-validation (accessed 2022-10-10).
(12) 21 CFR 820: Quality System Regulation for Medical Devices; Food and Drug Administration: Rockville, MD, 1996. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820 (accessed 2022-10-10).
(13) 21 CFR 11: Electronic Records; Electronic Signatures, Final Rule, in Title 21; Food and Drug Administration: Washington, DC, 1997. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-11 (accessed 2022-10-10).
(14) ISO 13485: 2016: Medical Devices -- Quality Management Systems -- Requirements for Regulatory Purposes; International Standards Organization: Geneva, 2016. https://www.iso.org/standard/59752.html#:~:text=ISO%2013485%3A2016%20specifies%20requirements,customer%20and%20applicable%20regulatory%20requirements. (accessed 2022-10-10).
(15) Okutan, O. Why the CSA Guidance is Not Progress and Undermines FDA’s Harmonisation Efforts within Medical Technology. Online article, LinkedIn, 2022. https://www.linkedin.com/feed/update/urn:li:activity:6984560426529218560/ (accessed 2022-10-10).
(16) EudraLex - Volume 4: Good Manufacturing Practice (GMP) Guidelines, Annex 11 Computerised Systems; European Commission: Brussels, 2011. https://health.ec.europa.eu/system/files/2016-11/annex11_01-2011_en_0.pdf (accessed 2022-10-10).
(17) Burgess, C.; Witzel, M. L. J.; Roussel, J.-M.; Quattrocchi, O.; Ermer, J.; Slabicky, R.; Martin, G. P.; Vivó-Truyols, G. Analytical Instrument and System (AIS) Qualification to Support Analytical Procedure Validation over the Life Cycle. Pharmacopoeial Forum 2022, 48 (1). https://www.uspnf.com/pharmacopeial-forum/pf-table-contents-archive
(18) ISO 29119-1:2013: Software and Systems Engineering — Software Testing — Part 1: General Concepts; International Standards Organization: Geneva, 2013. https://www.iso.org/standard/45142.html (accessed 2022-10-10).
(19) Bach, J. How Not to Standardize Testing (ISO 29119); blog post at Satisfice.com. https://www.satisfice.com/blog/archives/1464 (accessed 2022-10-10).
(20) Association for Software Testing. The ISO29119 Debate; online post, Sept 5, 2014. https://associationforsoftwaretesting.org/2014/09/05/the-iso29119-debate/ (accessed 2022-10-10).
(21) ISO/IEC 29119. Wikipedia. https://en.wikipedia.org/wiki/ISO/IEC_29119 (accessed 2022-10-10).
(22) McDowall, R. D. Are Spreadsheets a Fast Track to Regulatory Non-Compliance? LCGC Europe 2020, 33 (9), 468–476. https://www.chromatographyonline.com/view/are-spreadsheets-a-fast-track-to-regulatory-non-compliance
(23) McDowall, R. D. Spreadsheets: A Sound Foundation for a Lack of Data Integrity? Spectroscopy 2020, 35 (9), 27–31. https://www.spectroscopyonline.com/view/spreadsheets-a-sound-foundation-for-a-lack-of-data-integrity-
(24) ASTM e2500 Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment; American Society for Testing and Materials: West Conshohocken, PA, 2020. https://www.astm.org/e2500-20.html (accessed 2022-10-10).
(25) 21 CFR 211 - Current Good Manufacturing Practice for Finished Pharmaceuticals. Fed. Regist. 1978, 43 (190), 45014–45089. https://www.fda.gov/files/drugs/published/Federal-Register-43-FR-45077.pdf (accessed 2022-10-10).
(26) ISPE. GAMP Good Practice Guide: IT Infrastructure Control and Compliance, 2nd ed; International Society for Pharmaceutical Engineering: Tampa, FL, 2017.
(27) 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceuticals; Food and Drug Administration: Silver Spring, MD, 2008. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-211 (accessed 2022-10-10).
(28) Burgess, C.; Slabicky, R.; Quattrocchi, O.; Weitzel, M. L. J.; Roussel, J.-M.; Ermer, J.; Vivó-Truyols, G.; Stoll, D.; Botros, L. L. Analytical Instrument and System (AIS) Qualification: The Qualification Life Cycle Process. Pharmacopoeial Forum 2022, 48 (5). https://www.uspnf.com/pharmacopeial-forum/pf-table-contents-archive


R.D. McDowall is the director of R.D. McDowall Limited and the editor of the “Questions of Quality” column for LCGC Europe, Spectroscopy’s sister magazine. Direct correspondence to: SpectroscopyEdit@MMHGroup.com ●











Synthesizing Synthetic Oligonucleotides: An Interview with the CEO of Oligo Factory
February 6th 2024LCGC and Spectroscopy Editor Patrick Lavery spoke with Oligo Factory CEO Chris Boggess about the company’s recently attained compliance with Good Manufacturing Practice (GMP) International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Expert Working Group (Q7) guidance and its distinction from Research Use Only (RUO) and International Organization for Standardization (ISO) 13485 designations.



