Data Transforms in Chemometric Calibrations, Part 4A: Continuous-Wavelength Spectra and Discrete-Wavelength Models
In this column and its successor, we describe and explain some algorithms and data transforms beyond those commonly used. We present and discuss algorithms that are rarely, if ever, used in practice, despite having been described in the literature. These comprise algorithms used in conjunction with continuous spectra, as well as those used with discrete spectra.
We’ve previously published columns to present the common algorithms used for spectroscopic analysis, usually with an emphasis on near-infrared (NIR) quantitative analysis, although many algorithms were included to represent other types of spectroscopy (1,2). Those columns were broad in scope, but somewhat narrower in detail (we can’t include everything about everything, after all!). In this column (and possibly the next one or two), we present more details about some algorithms that are rarely seen or used, but deserve more attention than they have previously been accorded. We hope that by making information about them available, some scientists will be piqued to investigate them further, or at least try them out and compare results obtained from them with the results from the more common or more popular algorithms.
Our previous columns about data transforms (3,4) discussed data transformations that were intended to enhance calibration performance results based on spectra representing a small number of discrete (non-continuous) wavelengths, most commonly implemented through the use of a small number of interference filters. That class of data analysis algorithms was initially developed because early NIR spectrometer designs were often based on the use of discrete interference filters to select the wavelengths at which measurements of optical energy would be performed. While we digressed from that theme for a few columns, we always intended to return to it, and with this column we carry out that intention. While there is a measurement technology (the use of tilting filters; see [5]) that represents an intermediate case (tilting an interference filter enables the filter to pass a small range of wavelengths near the nominal wavelength [the designated wavelength it passes when perpendicular to the optical beam]; see page 82 in [6] for a graphical explanation, or [5] for the mathematics behind it all). In most applications, optical data is collected either discretely (as mentioned above) or “quasi-continuously” (that is, collected at closely spaced wavelengths over the range of wavelengths representing the spectrum).
We began this set of columns by discussing data transforms suited to calibrations based on spectral data at discrete wavelengths, and we will eventually return to those types of transforms, but now we interrupt that exposition in order to discuss data transforms better suited to continuous spectra. We believe that those are of more interest to our readers than the others, for the simple reason that the corresponding algorithms are in more common use. We will, however, eventually go back to finish our discussion of data transforms for discrete data; there are some enormously interesting and important things to learn about them, including the fact that “improving the performance” includes the case where the performance being improved is that of a second instrument using the same calibration model. In other words, while often discussed separately (as we’ve done ourselves [7,8]), “calibration transfer” is (in one sense, at least “simply”) another application of the underlying concept of modifying a data set to achieve a specific goal for the calibration model. In some cases, the modification of the data is incorporated into, or indeed consists of, modification of the calibration algorithm. Sometimes, it’s hard to tell the difference, so here we will not attempt to make the distinction, but rather discuss the various methodologies that have been applied to the problem without further categorizing them. Where we have discussed a particular approach in a previous column, we will simply reference that column and, if appropriate, include any further explanations we feel necessary to understanding the modeling procedure. A summary of these methods has been presented at the 2020 Eastern Analytical Symposium (9), and some of our discussion here is extracted from that presentation.
Hardware Approach
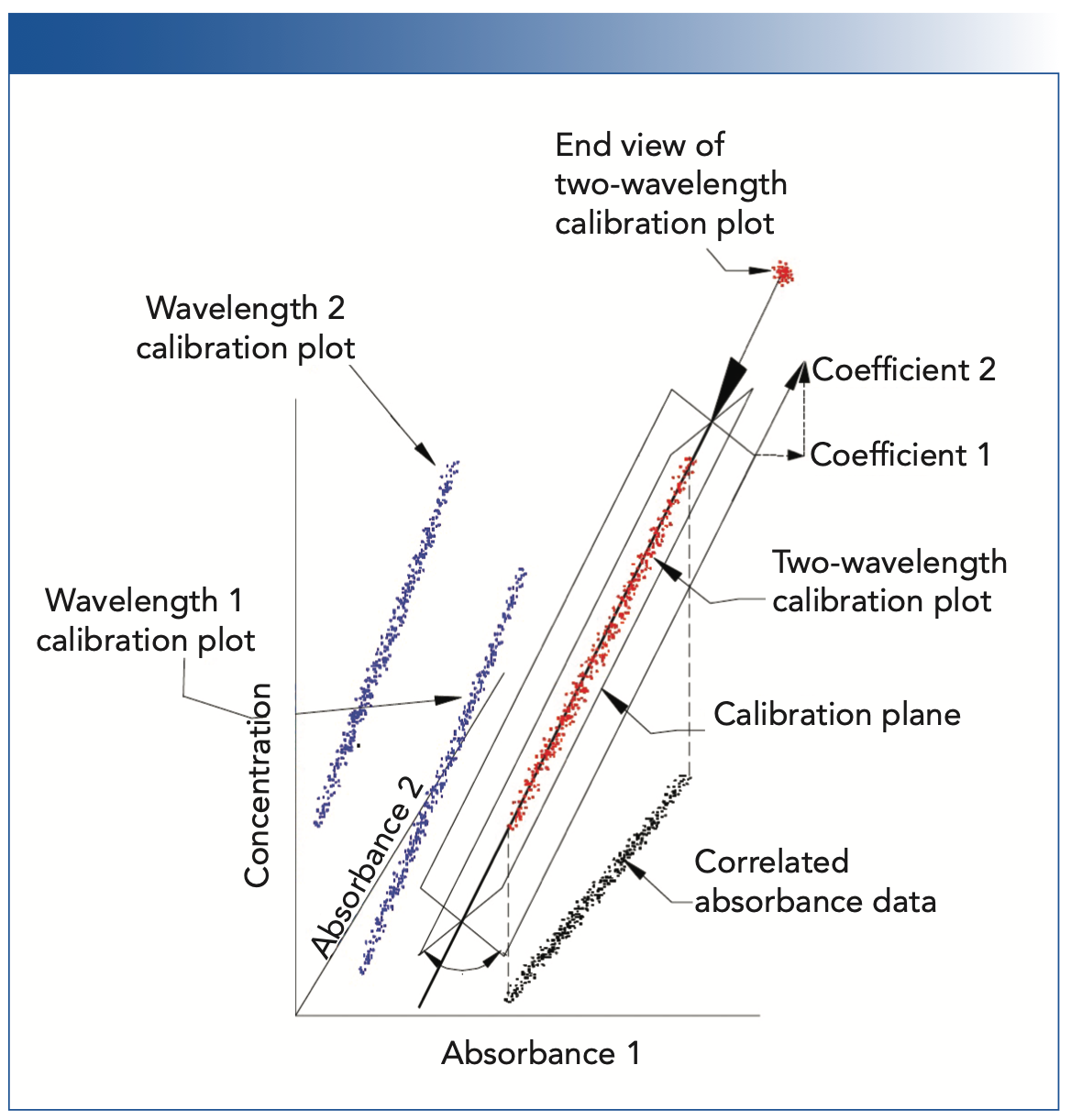
The hardware approach consists of matching instruments so well that the same calibrations would be developed on any of them individually. This is one of the topics that has been discussed previously in a recent series of our columns (10–14). The difficulty that arises is that the inevitable presence of noise in the spectral data, combined with the inherent tendency for the various wavelengths in the NIR spectra of a data set to be highly correlated, creates a problem. This problem is illustrated in one of our relatively recent columns (3). Figure 1 of that column, on page 18 of that issue and reproduced as Figure 1 in this column, displays how when spectral data at two (or more) wavelengths are inter-correlated, the three-dimensional (and higher-dimensional) relationships are circularly symmetric. Because of this effect, the calculated calibration plane (or hyperplane) is rotationally unstable around the axis of the higher-dimensional figure, and small changes in the error structure of the data can cause large changes in the orientation of the calibration plane; therefore, the coefficients representing that calibration plane are similarly unstable. Thus, no matter how well the hardware is matched, the inherent residual noise of the measurement process will make the calibration models appear to be different, even though this is only due to the irreducible noise of the measurement process. This instability of the calibration model was the bane of the early NIR developers, and led to many attempts to avoid or minimize it. These attempts gave rise to the software developments that are the forerunners of several of the modern data transformations still in use today.

![FIGURE 1: Illustration of the effect of intercorrelation of the calibration data on the determination of the calibration coefficients (reproduced from [3]).](/_next/image?url=https%3A%2F%2Fcdn.sanity.io%2Fimages%2F0vv8moc6%2Fspectroscopy%2F7d40df7ebf6b79c6c35ce53a8af95f2a130464b3-1168x1222.png%3Ffit%3Dcrop%26auto%3Dformat&w=3840&q=75 "FIGURE 1: Illustration of the effect of intercorrelation of the calibration data on the determination of the calibration coefficients (reproduced from [3]).")
FIGURE 1: Illustration of the effect of intercorrelation of the calibration data on the determination of the calibration coefficients (reproduced from [3]).
Software Approach
The software approach consists of algorithms that create the same models from similar data. The instability of the modeling process described above was not understood or appreciated for a long time. Other problems also afflicted the model development process, and it was often unclear which underlying effect was creating calibration difficulties in any particular case. One of the key problematic phenomena was the fact that most sample types of interest in the early days were (and still are, for that matter) solids, but not solid blocks of the material comprising the sample. Rather, the sample often consisted of fine powders composed of the sample material ground to a collection of particles that, with rare exceptions, could be characterized as being comprised of a mixture of particles in the size range from dust to sand. This created a problem in that when the sample is presented to an instrument, there is a large random component in what the instrument “sees.” If such a sample is removed from the holder used to contain the sample, presented to the instrument, then replaced in the holder, different facets of the individual particles of sample interact differently with the NIR radiation and thereby produce a different set of measured values of reflectance at the various wavelength used for the same sample. Examples of the “Particle Size Effect” (sometimes also called the “Repack Effect”) abound in the literature; several examples can be found in various editions of the Handbook of Near-Infrared Analysis, for example (6).
There was little the average scientist could do to directly affect either of these uncontrollable variables. What was possible, however, was to attempt devising algorithms that, as part of the calibration procedure, could empirically correct or compensate for the uncontrollable fluctuations. Many such algorithms were developed over the years, and we include descriptions of several of them in this (and our subsequent) column. We have already presented some of these algorithms, intended for use in conjunction with discrete wavelengths in predecessor columns (3,4) in this series. Now, we will begin describing algorithms intended for use in conjunction with wavelengths measured quasi-continuously over a range (spectra).
Note that while we list some of the algorithms here under particular headings, that listing reflects the fact that there may be a theoretical connection between the algorithm and the phenomenon it is being used to correct or compensate for. In practice, various scientists have used one or more of these algorithms to improve their calibration results in the face of any of the problem areas affecting their NIR calibrations. Sometimes, these transforms are used completely empirically to improve calibrations, even when the cause of the difficulty is unknown. An unfortunate side effect of this concept, verging on misuse of the algorithms, is the tendency of some scientists to attribute any improvement in the performance of a calibration (using the proposed data transform) to the effect of that proposed data transform. The possibility of other differences in the two cases (with and without the application of the proposed data transform) is not even considered. The possibility of other effects on the data, even a different contribution of the inevitable random noise, is completely ignored. Thus, while that approach may solve an immediate problem, it is lousy (although not quite junk) science.
We now present several algorithms that have been proposed by various scientists to mitigate the problems encountered when performing NIR calibration studies. Some of these were generated in the early days of chemometrics applied to NIR analysis. Indeed, some of them predate the development of NIR analysis itself and even of chemometrics. Some were developed by mathematicians who were using multivariate methods to solve problems in physics, chemistry and other sciences. Of course they did not call it “chemometrics;” it was done under the umbrella of the branch of mathematics called “statistics.” But the math was the same, regardless of the label.
With the advent of the modern chemometric algorithms, those initial early methods quickly fell into disuse and, to some extent, into disrepute. They came to be surrounded by an aura of being “primitive,” “old-fashioned,” not being up to modern standards, possibly incapable of solving modern problems. Maybe, but that’s unfortunate. Scientists in those days had plenty of difficult and complicated problems to solve, and they solved them. Without having a computer and a “magic” algorithm handy to do their thinking for them, they had to study the systems causing their difficulties and learn to understand them—to know what was going on “under the hood,” so to speak, so they could make intelligent guesses as to when and why they failed and how to make corrections. That sort of thinking, however,has become somewhat of a “lost art” nowadays, mainly the lost art of thinking about what we’re doing, and replacing thinking with computer processing. The only case we know of in recent times that reflects the historic approach is one that we discuss below: the work of Karl Norris.
In our list here, we include some of those older algorithms. Just because they’ve fallen into disuse does not mean that they are useless. Indeed, by avoiding being overly complicated, some of them are easier to use and to understand, both conceptually and by their effect on calibration results. We start with the simpler ones and move on to more advanced algorithms. Some of these manipulations are, arguably, not so much data transformations as they are modifications of the calibration algorithm. Being by far the most complicated and difficult-to-understand, we do not include the principal component regression (PCR) and partial least squares (PLS) algorithms here. We, as well as others, have presented and discussed both of those algorithms previously, in earlier columns of our series.
CLS (Classical Least Squares)
The classical least squares (CLS) algorithm is arguably the oldest algorithm used for spectroscopic calibration, being developed directly from the application of Beer’s law. In the earliest days, when both the instruments and the mathematics available were still very primitive, calibrating a spectroscopic instrument was more akin to calibrating a thermometer than a spectrometer. A single wavelength would be chosen. The scientist involved would develop a univariate relationship between the absorbance values of the sample at that wavelength, versus a measure of concentration of the desired analyte. In those days computers were not available either, and a “calibration” would often consist of a graph manually drawn on graph paper. We’ve discussed CLS in a series of previous columns starting with (15), another series beginning with (16), and a book on the topic (17). There were numerous limitations of this calibration method, not the least of which was the fact that it could be applied only to a clear liquid sample containing a single absorbing analyte in a non-absorbing solvent. As soon as a second absorbing material was added to the solution, it would interfere with the measurement of the intended analyte. In this case, a second measurement at a different wavelength would enable a correction to be made for the interfering material (17). In this case, it might then even be possible to measure the amount of both materials in the sample.
In principle, this process could be extended to more analytes in the sample (using three wavelengths to measure three absorbing ingredients in the mixtures, for example), but practical problems crop up. It’s still limited to liquid mixtures (at that time, at least). The ingredients must be noninteracting. For example, an attempt to apply this method to mixtures of water, ethanol, and acetic acid failed due to spectral distortions caused by hydrogen bonding (18). There are also questions about which wavelengths should be used and how should they be selected, among others. One solution to these difficulties was to use all the wavelengths in the spectrum. This algorithm became known in the spectroscopic community as the CLS algorithm, wherein the spectrum of a mixture of absorbing materials is considered as the sum of the absorbance spectra of the components of the mixture; the coefficients of the spectra of the ingredients represent the volume fractions of each of the various ingredients in the mixture. We’ve described and discussed this approach in a series of past columns (16,18–27), as well as it having been published in the formal literature (28). (Parenthetically, we note that, simultaneously, the multiple linear regression [MLR] algorithm became known to the spectroscopic community as the inverse least squares [ILS] algorithm; eventually, good sense prevailed, and the proliferation of terminology and abbreviations was cut short by the community’s decision that use of the term ILS was to be deprecated. The change in terminology does not affect the utility of the algorithms, however.)
Karl Norris’ Derivative-Ratio Technique for Reducing Spectral Data to One Wavelength
While the application of derivative transforms of spectral data (dnA/dλn, where n is generally between 1 and 4) has been in use almost as long as the technology of NIR analysis itself has been available, there is a growing recognition of the limitations of that data transform. Indeed, we ourselves recently published a critique of that data transform. One criticism is based on the fact that with digitized data, attempts to do so invariably result in an attempt to calculate 0/0, which is an error condition. Thus, one cannot actually calculate a derivative since that requires the wavelength difference between data points being used →0, a condition that cannot be achieved with digitized data. At best, the difference between adjacent digitized data points must be used for the calculation, as an approximation to the derivative. The behavior of the approximation under these conditions has been previously examined (29–33). Nevertheless, Karl Norris and Bill Hruschka very successfully programmed their computer to perform the calculations needed to implement calibrations using “derivative ratios,” which are the quotients of the derivative approximations. This program would also vary the wavelengths at which these various “derivatives” and their quotients were calculated, then perform the calibration calculations using the resulting “derivative” quotients instead of the original absorbance values. The program enables the user to automatically select the wavelengths to use, and also has an interactive mode where the user can use his intuition to “guide” the wavelength search.
It was undoubtedly this capability that enabled Karl to routinely win the “Software Shootout” competition of the Chambersburg Conference (formally the International Diffuse Reflection Conference [IDRC], held every other year at Wilson College in Chambersburg, Pennsylvania), to the point where the conference organizers asked Karl to refrain from entering the competition just to give the other competitors a chance to win!
The heart of the algorithm was a two-step process: First, compute “derivatives” of the spectrum centered at all available wavelengths. Secondly, calculate the ratios (or quotients) of those derivatives at selected wavelengths. Those quotients were then used instead of the original spectral data in an MLR-type calculation to create the calibration model.
The original implementation of this algorithm was created on the computer in Karl’s U.S. Department of Agriculture/Agricultural Research Service (USDA/ARS) laboratory using a proprietary FORTRAN compiler. Since the work was all done under the auspices of the USDA/ARS, it was all in the public domain, and Karl intended to make his code available for free to any interested scientist, and especially to students. The use of FORTRAN, however, created a stumbling block to that intention since most modern (Windows-based) computers did not have compatible compilers. Karl therefore collaborated with David Hopkins to convert his and Bill’s FORTRAN programs to an equivalent set of MATLAB programs, which Karl and David also intended to make available. With the death of Karl and the sudden unexpected death of David, with the assistance of David’s wife, a consortium of their friends were able to “rescue” the MATLAB code, and intend to make it available as Karl and David intended, although the details of how this will be accomplished have not been worked out.
Post-Calibration Augmented-Components Methodology
During the period from the late 1990s through the early 2000s, David Haaland and coworkers, working at Sandia National Laboratories in Albuquerque, New Mexico, devised a most ingenious algorithm to improve calibration (strictly speaking, prediction) performance for aqueous solutions of urea, creatinine and sodium chloride (NaCl). It started with the realization that, when performing a spectroscopic calibration experiment, the “common knowledge” said that only the known and measured properties of the samples can be used for relating the spectroscopy to the chemistry (34–36). Other (unmeasured) properties (such as temperature, pH, or other chemical variations of the samples) that can potentially affect the measurement process are typically ignored. In this particular case, the unmeasured property of interest to these scientists is temperature. Since the measurements involved were made in aqueous solutions, the effect of temperature on the spectra of the samples can severely interfere with the desired spectrum of the analyte. In the author’s words, “The unmodeled spectral component…is the temperature of the solution, which is a relatively large source of unmodeled spectral interference in the near-infrared spectra of the unknown samples. Of course, temperature by itself does not have a spectrum, but temperature exerts its influence on the samples by modifying the spectra of the samples” (35). The authors then proceed to introduce the use of PLS to extract a “temperature” spectrum from their aqueous solutions. Similarly, the authors also hypothesized the possibility of determining and removing the effect on the spectra of uncontrollable variations in the instrument.
The authors then demonstrated the ability of adding the “spectrum” of the temperature changes to the set of principal components to improve the performance of their calibration model. They were then able to apply their concept to other calibration algorithms, in particular to the CLS algorithm. An interesting sidelight is how Haaland and Melgaard (35) and your current authors independently developed similar graphic presentations of the development of the CLS algorithm. Haaland and Melgaard (35), however, also extended the concept to include spectra of unknown interferences. Since the CLS algorithm can be inverted and used to reconstruct the spectra of the mixtures and also of the mixture components, the difference between the reconstructed spectrum of a mixture and the actual measured spectrum of that mixture represents the “spectrum” of the unmeasured and previously unmodeled properties of the mixtures as described in their earlier paper (34). It is then a relatively straightforward exercise to add the “spectra” of the previously undetermined interfering phenomena (such as, for example, temperature) to the calibration model to correct for those interferences, without the need to re-measure the spectra of all the samples. This new algorithm was designated as the prediction-augmented classical least squares (PACLS) algorithm.
Haaland and coauthors then further extended their concept in two directions: 1) to produce a “hybrid” algorithm of CLS + PLS + PACLS, and 2) to expand the applicability of these new algorithms to maintenance, as well as development of spectroscopic calibrations. We don’t have space here to describe all these developments in detail, but an interested reader can easily find more information about them in the literature (34,35). Nevertheless, we note that these authors made a major contribution to the conceptual basis for various chemometric algorithms, particularly the use of PCR and PLS.
To go back into mathematical history, we find that mathematicians Brook Taylor and Jean-Baptiste Joseph Fourier (yes, he of the Fourier transform) developed what we now call Taylor series and Fourier series, respectively. What these two mathematical constructs have in common is their ability to approximate any arbitrary (although “well-behaved”) function to any desired degree of precision by including sufficiently many basis functions in the calculation (the “basis functions” are polynomials in the case of Taylor series, and trigonometric functions [sines and cosines] in the case of Fourier series). Other mathematicians (for example, Bessel and Legendre) demonstrated that other mathematical constructs had that same capability, to reconstruct (approximate to any desired degree of precision) any mathematical function through the use of a suitably chosen set of basis functions.
There are several more algorithms that are interesting and potentially useful, and that may be beneficial to an NIR practitioner who wants to investigate different ways to analyze data and produce superior calibration models. We will discuss some of these in a forthcoming Part B to this column.
A final word: If any of our readers are, or become, aware of the appearance of any of these algorithms, or the ones to be published on our next column, in the literature, we would appreciate your letting us know about those appearances, ideally with a full literature citation.
References
(1) Workman, J.; Mark, H. One Real Challenge That Still Remains in Applied Chemometrics. Spectroscopy 2020, 35 (6), 28–32.
(2) Workman, J.; Mark, H. Survey of Key Descriptive References for Chemometric Methods Used for Spectroscopy: Part II. Spectroscopy 2021, 36 (10), 16–19.
(3) Mark, H.; Workman, J. Data Transforms in Chemometric Calibrations: Application to Discrete-Wavelength Models, Part 1: The Effect of Intercorrelation of the Spectral Data. Spectroscopy 2022, 37 (2), 16–18,54.
(4) Mark, H.; Workman, J. Data Transforms in Chemometric Calibrations: Simple Variations of MLR, Part 1. Spectroscopy 2022, 37 (7), 14–19.
(5) Lofdahl, M. G.; Henriques, V. M. J.; Kiselman, D. A Tilted Interference Filter in a Converging Beam. Astron. Astrophys. 2011, 533, A82. DOI: 10.1051/0004-6361/201117305
(6) Ciurczak, E.; Igne, B.; Workman, J.; Burns, D. Handbook of Near Infrared Analysis, 4th ed.; CRC Press, 2020, 223.
(7) Workman, J.; Mark, H. Calibration Transfer Chemometrics, Part I: Review of the Subject. Spectroscopy 2017, 32 (10), 18–24.
(8) Workman, J.; Mark, H. Calibration Transfer Chemometrics, Part II: A Review of the Subject. Spectroscopy 2018, 33 (6), 22–26.
(9) NYSAS 2020 Gold Medal Award Presentation, EAS Virtual Meeting, November 18, 2020.
(10) Workman, J.; Mark, H. Calibration Transfer, Part II: The Instrumentation Aspects. Spectroscopy 2013, 28 (5), 12–21.
(11) Workman, J.; Mark, H. Calibration Transfer, Part III: The Mathematical Aspects. Spectroscopy 2013, 28 (6), 28–35.
(12) Workman, J.; Mark, H. Calibration Transfer, Part IV: Measuring the Agreement Between Instruments Following Calibration Transfer. Spectroscopy 2013, 28 (10), 24–33.
(13) Mark, H.; Workman, J. 2014 Review of Spectroscopic Instrumentation Presented at Pittcon. Spectroscopy 2014, 29 (5), 18–27.
(14) Workman, J.; Mark, H. Calibration Transfer, Part VI: The Mathematics of Photometric Standards Used for Spectroscopy. Spectroscopy 2014, 29 (11), 14–21.
(15) Mark, H.; Workman, J. More About CLS, Part 1: Expanding the Concept. Spectroscopy 2019, 34 (6), 16–24.
(16) Mark, H.; Workman, J. Classical Least Squares, Part I: Mathematical Theory. Spectroscopy 2010, 25 (5), 16–21.
(17) Mark, H. Principles and Practice of Spectroscopic Calibration; John Wiley & Sons, 1991.
(18) Mark, H.; Workman, J. Classical Least Squares, Part II: Mathematical Theory Continued. Spectroscopy 2010, 25 (6), 20–25.
(19) Mark, H.; Workman, J. Classical Least Squares, Part III: Spectroscopic Theory. Spectroscopy 2010, 25 (10), 22–31.
(20) Mark, H.; Workman, J. Classical Least Squares, Part IV: Spectroscopic Theory Continued. Spectroscopy 2011, 26 (2), 26–33.
(21) Mark, H.; Workman, J. Classical Least Squares, Part V: Experimental Results. Spectroscopy 2011, 26 (5), 12–22.
(22) Mark, H.; Workman, J. Classical Least Squares, Part VI: Spectral Results. Spectroscopy 2011, 26 (6), 22–28.
(23) Mark, H.; Workman, J. Classical Least Squares, Part VII: Spectral Reconstruction of Mixtures. Spectroscopy 2011,26 (10), 24–31.
(24) Mark, H.; Workman, J. Classical Least Squares, Part VIII: Comparison of CLS Values with Known Values. Spectroscopy 2012, 27 (2), 22–34.
(25) Mark, H.; Workman, J. Classical Least Squares, Part IX: Spectral Results from a Second Laboratory. Spectroscopy 2012, 27 (5), 14–19.
(26) Mark, H.; Workman, J. Classical Least Squares, Part X: Numerical Results from the Second Laboratory. Spectroscopy 2012, 27 (6), 28–35.
(27) Mark, H.; Workman, J. Classical Least Squares, Part XI: Comparison of Results from the Two Laboratories Continued, and Then the Light Dawns. Spectroscopy 2012,27 (10), 12–17.
(28) Mark, H.; Rubinovitz, R.; Dahm, D.; Dahm, K.; Heaps, D.; Gemperline, P. Pittcon 2011, Atlanta, GA, 2010 (pub).
(29) Mark, H.; Workman, J. Derivatives in Spectroscopy, Part I: The Behavior of the Derivative. Spectroscopy 2003, 18 (4), 32–37.
(30) Mark, H.; Workman, J. Derivatives in Spectroscopy, Part II: The True Derivative. Spectroscopy 2003, 18 (9), 25–28.
(31) Mark, H.; Workman, J. Derivatives in Spectroscopy, Part III: Computing the Derivative. Spectroscopy 2003,18 (12), 106–111.
(32) Mark, H.; Workman, J. Chemometrics Derivatives in Spectroscopy, Part IV: Calibrating with Derivatives. Spectroscopy 2004, 19 (1), 44–51.
(33) Mark, H.; Workman, J. Chemometrics in Spectroscopy: Derivatives in Spectroscopy Update. Spectroscopy 2004, 19 (11), 110–112.
(34) Haaland, D. M. Synthetic Multivariate Models to Accommodate Unmodeled Interfering Spectral Components During Quantitative Spectral Analyses. Appl. Spectrosc. 2000, 54 (2), 246–254.
(35) Haaland, D. M.; Melgaard, D. K. New Prediction-Augmented Classical Least-Squares (PACLS) Methods: Application to Unmodeled Interferents. Appl. Spectrosc. 2000, 54 (9), 1303–1312.
(36) Haaland, D. M.; Melgaard, D. K. New Classical Least-Squares/Partial Least-Squares Hybrid Algorithm for Spectral Analyses. Appl. Spectrosc. 2001, 55 (1), 1–8.
About the Authors


Howard Mark serves on the Editorial Advisory Board of Spectroscopy, and runs a consulting service, Mark Electronics, in Suffern, New York. Direct correspondence to: SpectroscopyEdit@mmhgroup.com ●



Jerome Workman, Jr. serves on the Editorial Advisory Board of Spectroscopy and is the Senior Technical Editor for LCGC and Spectroscopy. He is the co-host of the Analytically Speaking podcast and has published multiple reference text volumes, including the three-volume Academic Press Handbook of Organic Compounds, the five-volume The Concise Handbook of Analytical Spectroscopy, the 2nd edition of Practical Guide and Spectral Atlas for Interpretive Near-Infrared Spectroscopy, the 2nd edition of Chemometrics in Spectroscopy, and the 4th edition of The Handbook of Near-Infrared Analysis. ●
















