Determination of the Relative Prevalence of Lurasidone Metabolites in Urine Using Untargeted HRMS
Special Issues
For lurasidone treatment adherence testing, an untargeted high-resolution mass spectrometry method was employed, using known positive human urine samples to identify the lurasidone metabolites and their relative abundance in urine.
Lurasidone is an atypical antipsychotic that was approved by the FDA in 2010 to treat bipolar depression and schizophrenia. Like other antipsychotics, adherence to lurasidone is critical for successful disease treatment. Thus, therapeutic drug monitoring (blood testing) is often employed by clinicians to monitor adherence. Urine drug testing, with its advantages over blood testing, is another method used to confirm medication adherence. However, analytes used in blood testing are often very different than those used for testing in urine, where nonactive metabolites are often most prevalent. Choosing metabolites in urine that are relatively prevalent affords optimal method sensitivity, and thus improved testing results for adherence. To ensure optimal lurasidone adherence testing, an untargeted high-resolution mass spectrometry method was employed, using known positive human urine samples to identify the lurasidone metabolites and their relative abundance in urine. This testing identified a different primary urine metabolite from what has been reported in blood. The higher prevalence of this metabolite will improve lurasidone urine adherence monitoring.
Medication monitoring has become increasingly important for successful treatment of patients with mental health diseases because adherence to treatment is generally poor, especially in the schizophrenic population (1–8). Urine has become an alternative to blood or plasma medication monitoring due to its noninvasive nature and ease of collection. Whereas blood or plasma drug testing usually involves the identification and quantitation of the parent compound or active metabolites, or both, the success of urine drug testing (UDT) is largely dependent on analysis of any metabolites of the parent compound. Although the parent compounds may be present in urine, often they are at very low concentrations relative to metabolites, and therefore do not provide the sensitivity required for medication monitoring. Urine metabolites are often predicted from identification in blood, plasma, or specific testing methods, such as gas chromatography-mass spectrometry (GC-MS), extractions, radioactivity, and using in vitro or animal samples. However, it has been shown that these methods are not always successful in identifying the most abundant urinary metabolite (9–12). Without suitable metabolites to test, a negative UDT result could prompt a clinician to alter treatment for a patient when treatment need not be altered. Therefore, a generic, untargeted approach is useful for the successful identification of urinary metabolites suitable for highly sensitive medication monitoring. Liquid chromatography–high resolution mass spectrometry (LC–HRMS) provides a sensitive and nonspecific detection method for setting up such an experiment.
Lurasidone (Latuda) is an atypical antipsychotic that was approved for the treatment of acute symptoms of schizophrenia (13,14) and bipolar depression (15,16) in 2010 and 2013, respectively. It is commercially available as 20 mg, 40 mg, 60 mg, 80 mg, and 120 mg tablets, and is typically prescribed or administered at 40 or 80 mg per day. It is absorbed after oral administration with a bioavailability of 9–19%. Dosing is designed to be with food, which can increase the bioavailability by 100%. The mean elimination half-life is 18 h. Steady state serum concentrations for lurasidone are typically achieved after seven days of dosing (17–20). Lurasidone is metabolized in the liver primarily by CYP3A4. Metabolism includes oxidative N-dealkylation, hydroxylation of the norborane ring, S-oxidation, and reductive cleavage of the isothiazole ring, followed by S-methylation. Nearly two dozen metabolites of lurasidone have been previously identified, and only ~9% of the dose is excreted in urine (17–20). Typically, adherence to lurasidone therapy is monitored by evaluating levels of lurasidone and M11/ID-20219 (one of its metabolites) that were each predicted to be present in urine at approximately 12 and 24%, respectively. The structures for lurasidone and many of the confirmed metabolite structures can be seen in Table I.


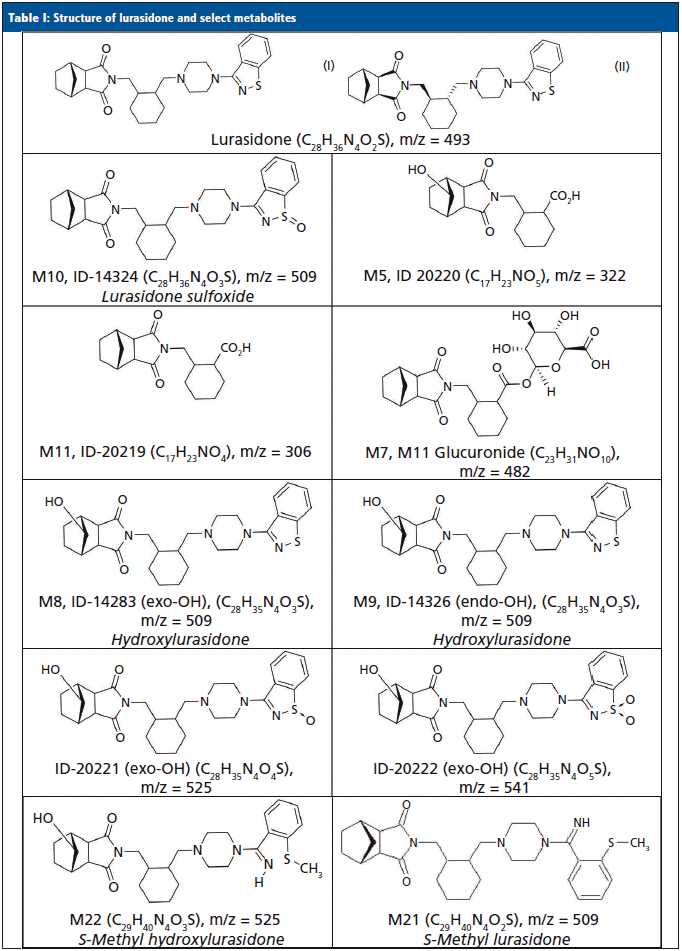
Previously, we reported the identification of novel metabolites for monitoring aripiprazole, brexpiprazole, haloperidol, and quetiapine in urine that were not originally predicted (9–12). Because there are some similarities of these antipsychotics to lurasidone, we decided to determine if the urinary lurasidone compound(s) predicted from plasma studies were indeed the most abundant prior to development of a confirmation method. This work reports the identification of lurasidone and prevalent lurasidone metabolites in urine using LC–HRMS from patients prescribed lurasidone. Additionally, confirmation of the most prominent metabolites was tested in a validated, targeted, quantitative liquid chromatography–tandem mass spectrometry method (LC–MS/MS), which are at odds with current reports of urine metabolites (17–20).
Experimental
Chemicals
Lurasidone, lurasidone-d8, and hydrocodone-d6 were purchased from Cerilliant (Round Rock, Texas). Hydroxylurasidone was a custom synthesis product purchased from 13C Molecular (Greensboro, North Carolina). All solvents, including methanol (optima grade), formic acid (88%), acetonitrile (optima grade), ammonium acetate (optima grade), and isopropanol (optima grade), were purchased from VWR (Radnor, Pennsylvania, USA). Drug-free human urine was acquired from UTAK Laboratories (Valencia, California). Standards for S-methyl lurasidone and S-methyl hydroxylurasidone were not commercially available, and synthesis requests were unsuccessful.
Sample Sets
Identification of lurasidone metabolites using LC–HRMS was completed on 13 authentic urine samples from patients who were prescribed the medication. After metabolite identification was complete, an LC–MS/MS confirmation was validated. An additional 56 patients were prescribed lurasidone at different doses, with specimens collected over three separate days for each patient used to confirm the accuracy of the method. These samples were provided voluntarily, and anonymously, to assist with the development of a lurasidone confirmation method. No identifying or demographic information was collected on these volunteers, other than the prescribed lurasidone dose. There was an alphanumeric code from the clinic that was provided to track the patients who provided samples over the course of the three separate days. None of the results were shared with the clinician to assist with treatment. Ameritox is accredited by the College of American Pathologists (CAP) and abides by CAP, Clinical Laboratory Improvement Amendments (CLIA), and Health Insurance Portability and Accountability Act (HIPAA) requirements. Due to the secondary analysis nature of this work and the absence of clinical conclusions, neither the United States Food and Drug Administration (FDA) nor other clinical trial review or approval was obtained by Ameritox. Writing this manuscript did not involve human subjects, as defined by the U.S. Code of Federal Regulations (45 CFR 46.102); thus, an Institutional Review Board (IRB) approval of these specific research activities was unnecessary.
LC–HRMS Sample
Preparation and Analysis
Thirteen patient urine specimens (100 µL) were diluted 5X with 400 µL of a reference standard, (0.25 µg/mL of hydrocodone-d6 in water). Hydrocodone-d6 was used as an internal reference standard for all LC–HRMS injections, to guarantee successful injection of the sample, and provide a retention time marker. Prepared samples were injected (5 µL) and separated on a Phenomenex Kinetex Phenyl-Hexyl, 2.1 x 50-mm, 2.6-µm column (Torrance, California) at 50 °C, and analyzed on an Agilent 6530 Q-TOF (quadrupole time-of-flight mass spectrometer) with an Agilent 1290 LC system (Santa Clara, California). The LC–QTOF method conditions are detailed in a previous publication (12). A lurasidone control in drug-free urine (75 ng/mL) was run, along with the patient samples, to assist in positive identification of the parent compound, if present. No other standards were available or purchased to assist in identification, until a confirmation method was developed. Each sample was injected and analyzed twice.
The MS-only data were processed using Agilent Mass Hunter Qualitative Analysis and PCDL (Personal Compound Database and Library) manager software. A database of lurasidone and 11 of its possible metabolites' chemical formulas (Table I) was compiled, and used to search against the samples. The software matched compounds based on retention time (if available), mass (±20 parts per million or ppm), the isotopic distribution pattern, and the isotopic spacing theoretically derived from the chemical formula. To be identified as positive and a potential lurasidone metabolite, a compound had to have consistent retention times across multiple patient samples when a known retention time was lacking; otherwise, the retention times had to be within ±0.05 minutes of a control. The mass accuracy had to be within ±20 ppm; and the composite score of the mass accuracy and isotopic features had to be ≥70 (out of a possible 100). Compounds that had the highest area counts were also ranked and noted as the most abundant. To assist with differentiation of structural isomers, such as M8/M9 and M10, fragmentation spectra were obtained and reviewed to identify which isomer was present at an identified metabolite peak, as needed.
LC–MS/MS Sample
Preparation and Analysis
Hydroxylurasidone was received as a neat solid that was dissolved into methanol at a concentration of 1 mg/mL, and lurasidone was received as a 100 µg/mL methanolic standard. Hydroxylurasidone and lurasidone were combined and diluted into a methanolic stock that was then further diluted into normal, drug-free human urine, to reach the appropriate calibrator (5, 25, 100, 500, and 1000 ng/mL) and quality control levels (75 ng/mL). Lurasidone-d8, 1 mg/mL methanolic stock, was diluted to 900 ng/mL in 0.1% formic acid in water solution. A 100 µL aliquot of the sample (patient sample, calibrator, or quality control stock) and 400 µL of lurasidone-d8 internal standard in 0.1% formic acid were added to a vial. Vials were then capped and vortexed for 10 s prior to injection of 5 µL.
Samples were analyzed by LC–MS/MS on a Waters Acquity UPLC Xevo TQ-MS system (Waters Corporation, Milford, Massachusetts), a Waters Acquity UPLC CSH Phenyl-Hexyl 2.1 x 50-mm, 1.7-µm UPLC column. The LC method and MS conditions can be found in Strickland and associates (12). Analyte transitions are listed in Table II. The acquisition method was run in dynamic multiple reaction monitoring (MRM) mode, in order to maximize the number of points across the various analyte peaks. The validation of this method followed CAP and CLIA guidelines (21–25), and an internal SOP (standard operating procedure) that has been described in detail elsewhere (26). It should be noted that, due to the lack of standards for S-methyl lurasidone and S-methyl hydroxylurasidone, they were unable to be validated, and estimates of their concentration were made by comparing the quantiative peak area ratio to the lurasidone calibration curve. These compounds were included as a proof of concept, to show their estimated prevalence and relative importance for lurasidone compliance in UDT for when standards might be available. Also, due to the lack of standards, the transition parameters were estimated from hydroxylurasidone and are not optimized.


Results
The metabolite identification from the 13 patients analyzed by LC-QTOF can be seen in Table III. Compounds identified with the highest confidence (>90%) are highlighted in green, while less confident (>70% but <90%) compounds are highlighted in yellow. The three most abundant compounds for each specimen and replicate are noted with a star-asterisk in the respective square. For unidentified or not confidently identified compounds (<70%) for a given specimen replicate, the field is blank. It is clear that, although lurasidone was identified in almost all of the samples, it was not consistently among the most abundantly identified compounds. Metabolite M11, the predicted major metabolite, was rarely confidently detected in these samples. Instead of lurasidone and M11, metabolites M21 (S-methyl lurasidone), M22 (S-methyl hydroxylurasidone), and isomer M8/M9 (hydroxylurasidone), or isomer M10 (lurasidone sulfoxide), were frequently detected. To determine whether hydroxylurasidone or the lurasidone sulfoxide (isomers) was present, the collected fragmentation data from the QTOF were analyzed, and are shown in Figure 1. The identification of a peak at m/z 182 (red circle in Figure 1) confirmed the isomer as hydroxylurasidone by indicating a fragmentation of the hydroxylated norborane ring. If the identity was the lurasidone sulfoxide, expected fragmentation peaks of m/z 152 or 237 from the oxidized sulfur atom on the isothiazole ring structure would be present. Additionally, the unhydroxylated norborane ring would have an expected m/z of 166. The absence of those expected peaks (m/z 152, 237, and 166) in the spectra confirms the identity of the metabolite as hydroxylurasidone, and a custom synthesis of the molecule was requested to validate a confirmation method. S-methyl lurasidone and S-methyl hydroxylurasidone were also requested as custom synthesis products, but attempts to synthesize for the method were unsuccessful.


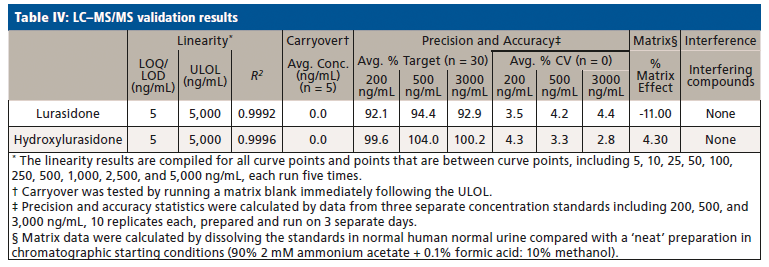
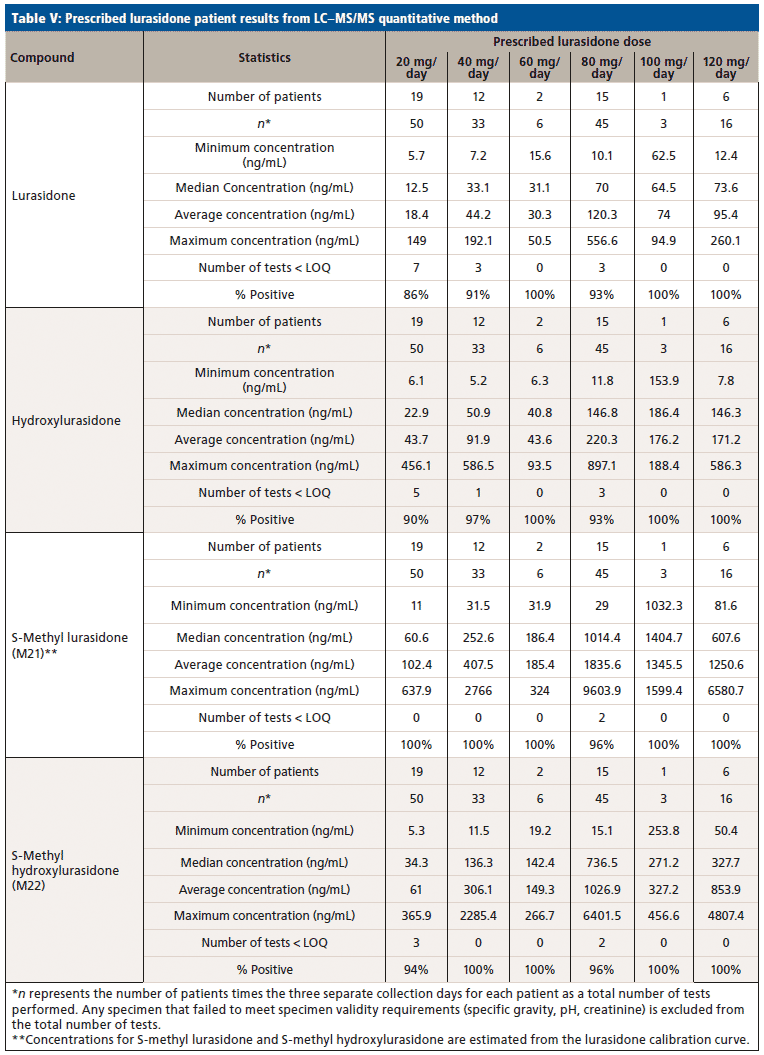
Upon receiving the hydroxylurasidone standard, an LC–MS/MS method was developed and validated. The results of validation of lurasidone and hydroxylurasidone are shown in Table IV. Although S-methyl lurasidone and S-methyl hydroxylurasidone were included in the method, without standards, validation was unable to be completed, and is the reason for their exclusion from Table IV. To ensure the ability to successfully detect and quantify lurasidone and hydroxylurasidone in patient specimens, samples from 56 additional patients (from three separate collection days) were provided for testing with the validated method. The results of these patient analyses are summarized in Table V, and separated by the prescribed dose. It is clear that testing for hydroxylurasidone helps with positive confirmation of taking lurasidone medication. It also appears, from the estimated concentrations of the S-methyl lurasidone and S-methyl hydroxylurasidone, that confirmation would be easier with these metabolites, because they are more abundant than both lurasidone and hydroxylurasidone. However, the lack of standards for these compounds makes it impossible to currently validate a method for reporting UDT results for these compounds.


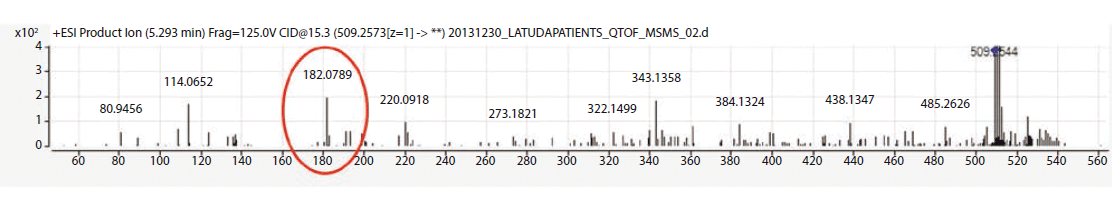
Figure 1: Fragmentation data from sample 8 Identified M8-M9-M10 Peak.
Discussion
The advantages of HRMS analysis have been reviewed in the literature, including the extreme selectivity of such methods (10–12,27–30). Using this method, authentic urine samples of human subjects who were known to be taking chronic doses of lurasidone were tested for the presence of lurasidone and 11 possible metabolites. Due to the high mass resolving power and low mass error on the QTOF, compounds that have similar mass to charge ratios, but different chemical formulas, were differentiated with the searching algorithm (hydroxylurasidone and S-methyl lurasidone). Also, by eliminating extraction preparation methods, compound loss was mitigated. Using liquid as opposed to gas chromatography also helped ensure that compounds with low volatility can still be accurately analyzed. Surprisingly, neither of the predicted major urinary metabolites M5 nor M11 were found to be consistently excreted through human urine in large detectable amounts. This result could be due to the fact that M5 and M11 both have a carboxylic acid moiety that would be a possible glucuronidation target. This was considered, and the glucuronidated versions were searched for in patient samples and poorly identified. While this may indicate that the glucuronide metabolites of M5 and M11 are not present in urine in any appreciable amount, it could also be due to the poor ionizability of glucuronidated compounds. To confirm, an additional hydrolysis study could be completed to see if there is an increase in the prevalence of M5 and M11 in patients after hydrolysis. However, with the significant presence of hydroxylurasidone (M8/M9), S-methyl lurasidone (M21), and S-methyl hydroxylurasidone (M22), it seemed unnecessary to pursue hydrolysis as a means for analyzing for lurasidone compliance.


The presence of these metabolites in urine was not well predicted from results in blood. Hydroxylurasidone was estimated to have a prevalence of 2.8% as the M8 isomer, and 0.4% as the M9 isomer; S-methyl lurasidone and S-methyl hydroxylurasidone were not predicted at any measurable amount (17–20). It does appear that the estimate of lurasidone at 12% might be reasonable, as all but one of the 13 patients in metabolite discovery had detectable amounts of the parent compound (17–20).
To better understand the relative amounts of each metabolite, lurasidone, hydroxylurasidone, S-methyl lurasidone, and S-methyl hydroxylurasidone present in urine, the results from the 56 patients used during method validation were analyzed. These results in Table V show that hydroxylurasidone, S-methyl lurasidone, and S-methyl hydroxylurasidone are present at approximately 2x, 7x, and 5x times, respectively, relative to lurasidone. All of the compounds show a general increase in concentration and percent positivity rate with increasing doses. It is clear that S-methyl lurasidone and S-methyl hydroxylurasidone provide slightly better positivity correlations at lower doses, but hydroxylurasidone does appear to provide enough benefit to help compensate for lower prevalence of lurasidone. Therefore, with the lack of available standards for S-methyl lurasidone and S-methyl hydroxylurasidone, hydroxylurasidone was validated to assist in UDT for lurasidone compliance.


Conclusion
We successfully identified prevalent lurasidone metabolites in urine. Based on those identifications, we successfully validated a method for the purpose of UDT monitoring of lurasidone. The hydroxylurasidone metabolite provides benefit for lurasidone UDT monitoring by being more prevalent in the urine than lurasidone by ~2x, and providing more consistent positivity correlation at lower lurasidone doses. Although other metabolites are present in the urine in large concentrations, standards for those compounds are not available at this time. However, the proof-of-concept work with S-methyl lurasidone and S-methyl hydroxylurasidone shows that they are ~5x and ~7x greater in abundance than lurasidone, respectively, and would provide even better positivity correlation at low doses.
References
(1) M. Ko, and T. Smith, Urine Drug Monitoring in Patients on Prescribed Antipsychotic Medications. 29th Annual US Psychiatric and Mental Health Congress Oct 21-24 (http://www.ingenuityhealth.com/wpcontent/uploads/2016/11/Mental_Health_Populations_Study_US_Psych_2016_Poster.pdf) (2016).
(2) A.E. Cooper, P. Hanrahan, and D.J. Luchins, Drug Benefit Trends 15(8), 34 (2003).
(3) C.R. Dolder, J.P. Lacro, L.B. Dunn, and D.V. Jeste, Am. J. Psychiatry 159, 103 (2002).
(4) S. Offord, J. Lin, D. Mirski, and B. Wong, I Adv. Ther. 30(3), 286 (2013).
(5) D.I. Velligan, F. Lam, L. Ereshefsky, and A.L. Miller, Psychiatr. Serv. 54, 665 (2003).
(6) D.I. Velligan, Y-W. F. Lam, D.C. Glahn, J.A. Barrett, N.J. Maples, L. Ereshefsky, and A.L. Miller., Schizophr. Bull. 32(4), 724–742 (2006).
(7) D.I. Velligan and P.J. Weiden, Psychiatr. Times 23(9), 1–2 (2006).
(8) R.A. Millet, P. Woster, M. Ko, M. DeGeorge, and T. Smith, Adherence to Treatment with Antipsychotic Medications Among Patients with Schizophrenia, Major Depressive Disorder, or Biopolar Disorder. Poster Presentation. (US Psychiatric and Mental Heal Congress (USPMHC) San Diego, CA,2015).
(9) J. McEvoy, R.A. Millet, K. Dretchen, A.A. Morris, M.J. Corwin, and P. Buckley, Psychopharmacology 231(23), 4421–4428 (2014).
(10) J.R. Enders, S.G. Reddy, E.C. Strickland, and G.L. McIntire, Clin. Mass. Spec. 6, 21–24 (2017).
(11) O.T. Cummings, E.C. Strickland, J.R. Enders, and G.L. McIntire, J. Anal. Toxicol. 42(4), 214–219 (2017).
(12) E.C. Strickland, O.T. Cummings, A,A. Morris, A. Clinkscales, and G.L. McIntire, J. Anal. Toxicol. 40(8), 687–693 (2016).
(13) M.P. Cruz, Drug Forecast 36(8), 489–492 (2011).
(14) S. Caccia, L. Pasina, and A. Nobili, Neuropsychiatric Disease and Treatment, 2012(8), 155–168 (2012).
(15) R. Franklin, S. Zorowitz, A.K. Corse, A.S. Widge, and T. Deckersbach, Neuropsychiatr.Dis. Treat. 2015(11), 2143–2152 (2015).
(16) M. Ostacher, D. Ng-Mak, P. Patel, D. Ntais, M. Schlueter, and A. Loebel, The World Journal of Biological Psychiatry, 19(8), 1–16 (2017). http://doi.org/10.1080/15622975.2017.1285050 (2017).
(17) Product Information: LATUDA oral tablets, lurasidone hydrochloride oral tablets. Sunovion Pharmaceuticals Inc. Marlborough, MA (2018).
(18) R.C. Baselt, Disposition of Toxic Drugs and Chemicals in Man (Biomedical Publications, Seal Beach, CA, 11th Ed., 2010), pp 1232–1233.
(19) Sunovion Pharmaceuticals Inc. Center for Drug Evaluation and Research Approval Package (Lurasidone). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/200603Orig1s010.pdf, Accessed May 8, 2018.
(20) T. Ishibashi, T. Horisawa, K. Tokuda, T. Ishiyama, M. Ogasa, R. Tagashira, K. Matsumoto, H. Nishikawa, Y. Ueda, S. Toma, H. Oki, N. Tanno, I. Saji, A. Ito, Y. Ohno, and M. Nakamura, J. Pharmacol. Exp. Ther. 334(1), 171–181 (2010).
(21) Association of Public Health Laboratories. CLIA-Compliant Analytical Method Validation Plan and Template for LRN-C Laboratories. December 2013.
(22) F.T. Peters, O.H. Drummer, and F. Musshoff, Forensic Sci. Int. 165, 216–224 (2007).
(23) B. Levine, Principles of Forensic Toxicology (AACC Press, Washington, DC, 2nd edition 2003) pp 114–115.
(24) U.S. Department of Health and Human Services, Food and Drug Administration. Guidance for Industry-Bioanalytical Method Validation (2001).
(25) National Laboratory Certification Program (NLCP). Manual for Urine Laboratories. October 2010.
(26) J.R. Enders and G.L. McIntire, J. Anal. Toxicol. 39, 662-667 (2015).
(27) J.M. Colby, K.L. Thoren, and K.L. Lynch, J. Anal. Toxicol. 42(4), 201–213 (2018).
(28) E. Partridge, S. Trobbiani, P. Stockham, T. Scott, and C.A. Kostakis, J. Anal. Toxicol. 42(4), 220–231 (2018).
(29) J.M. Colby,K.L. Thoren, and K.L. Lynch, J. Anal. Toxicol. 41(1), 1–5 (2017).
(30) S.K. Manier, A. Keller, J. Schaper, and M.R. Meyer, Nature: Sci. Rep. 9(2741), 1–11 (2019).
Erin C. Strickland is with Ameritox, LLC, in Greensboro, North Carolina. Jeffrey R. Enders is with the Molecular Education, Technology and Research Innovation Center of North Carolina State University, in Raleigh, North Carolina. Gregory L. McIntire is with Premier Biotech, in Minneapolis, Minnestota. Direct correspondence to: gregorymcintire2@gmail.com













LIBS Illuminates the Hidden Health Risks of Indoor Welding and Soldering
April 23rd 2025A new dual-spectroscopy approach reveals real-time pollution threats in indoor workspaces. Chinese researchers have pioneered the use of laser-induced breakdown spectroscopy (LIBS) and aerosol mass spectrometry to uncover and monitor harmful heavy metal and dust emissions from soldering and welding in real-time. These complementary tools offer a fast, accurate means to evaluate air quality threats in industrial and indoor environments—where people spend most of their time.


NIR Spectroscopy Explored as Sustainable Approach to Detecting Bovine Mastitis
April 23rd 2025A new study published in Applied Food Research demonstrates that near-infrared spectroscopy (NIRS) can effectively detect subclinical bovine mastitis in milk, offering a fast, non-invasive method to guide targeted antibiotic treatment and support sustainable dairy practices.

Smarter Sensors, Cleaner Earth Using AI and IoT for Pollution Monitoring
April 22nd 2025A global research team has detailed how smart sensors, artificial intelligence (AI), machine learning, and Internet of Things (IoT) technologies are transforming the detection and management of environmental pollutants. Their comprehensive review highlights how spectroscopy and sensor networks are now key tools in real-time pollution tracking.


