Do You Understand the FDA’s Updated Approach to Pre-Approval Inspections?
Spectroscopy
The Food and Drug Administration (FDA) has issued an updated version of its Compliance Policy Guide for Pre-Approval Inspections (PAI). This article will guide you on this policy’s impact on analytical procedures and data included within regulatory submissions.
The U.S. Food and Drug Administration (FDA) recently issued an updated version of its Compliance Policy Guide for Pre-Approval Inspections (PAI). If you are not aware of the impact on analytical procedures and data included in regulatory submissions, then please read on.
R D McDowall
One of the U.S. FDA’s Compliance Program Guides (CPG), numbered 7346.832, is entitled Pre-Approval Inspections. This document is used to guide and inform inspectors conducting this type of inspection before granting a license to market an active pharmaceutical ingredient (API) or medicinal product for the U.S. market. The Able Laboratories fraud case in 2005 was a turning point for this document. The Agency had conducted seven PAIs of Able and not found any evidence of falsification. A whistle-blower in the quality-control (QC) laboratories at Able then informed the local FDA field office of the data integrity issues in the company. The following day, a posse of inspectors rode into town and camped at the Able facility looking at electronic data and comparing these records with the printouts that had been the focus of earlier inspections. Their findings are summarized in the 483 observations issued in July 2005 that resulted in the company’s going bankrupt. The Able 483 is still available on the FDA web site (1).
Updating CPG 7346.832, Take 1: May 2010
The FDA realized that their inspection approach of paper review was inadequate, and that inspections had to focus on the electronic records in laboratory computerized systems. As a result, paper records became incidental to the inspection of computerized systems. This required rewriting the existing CPG 7346.832 (yes, there is a “Star Trek” fan working at the FDA!), and the new version was issued in May 2010 and became fully effective in May 2012 (2). This gap allowed time for industry to understand the new inspection approach, as well as time to train inspectors on ways for inspecting electronic records within the various data systems used in regulated GMP laboratories.
The key changes in the rewritten version were the three inspection objectives (2):
- Objective 1: Readiness for Commercial Manufacturing
- Objective 2: Conformance to the Application
- Objective 3: Data Integrity Audit.
Reading this list of objectives, one could be forgiven for assuming that the only data integrity focus was objective 3. However, you would be mistaken; data integrity runs throughout all three objectives. This inspection approach, coupled with how to inspect electronic systems, has been used since 2012, and as a result there have been an increased number of 483 observations and warning letters issued to laboratories concerning data integrity.
Updating CPG 7346.832, Take 2: September 2019
September 2019 saw the issue of an updated version of CPG 7346.832 that still contains the three objectives, but the reason for the revision is stated on the front of the document as (3):
Program revised to add instructions for potential official action indicated (pOAI) reporting responsibilities and to align with the Center for Drug Evaluation and Research (CDER) and Office of Regulatory Affairs (ORA) agreement Integration of FDA Facility Evaluation and Inspection Program for Human Drugs: A Concept of Operations.
On the front of the document, the implementation date is September 16, 2019, with completion by September 16, 2022. Thus, the document is effective now, but I think that inspectors will need training in the additional scope and content, and therefore it will be fully effective later rather than sooner. Before we discuss the new CPG in more detail, we need to understand what a pOAI is. Readers who know the classification of 483 observations can skip the next section.
Understanding a Form 483 Inspectional Observation
To the uninitiated, let me take a short detour to explain how the Agency classifies non-compliances on an FDA Form 483–Inspectional Observations. The completed Form 483 (typically shortened to a 483) is left with senior management at the close out meeting of an FDA investigation, and it lists the non-compliances found during an inspection. The inspector does not classify them at the closing meeting; classification is undertaken after the inspection, and is subject to Agency review that takes into account each non-compliance, its seriousness, and compliance history of the inspected organization.
There are three classifications that can be given by the FDA to each 483 observation; these can be found in the exotically titled Field Management Directive 86 (FMD 86) Establishment Inspection Report Conclusions and Decisions (4):
- NAI–No Action Indicated: No objectionable conditions or practices were found during the inspection, or the significance of the documented objectionable conditions found does not justify further FDA action.
- VAI–Voluntary Action Indicated: Objectionable conditions were found and documented, but the District and/or Center is not prepared to take or recommend any of the regulatory actions, because objectionable conditions do not meet the threshold for regulatory action. The district may request a written response from the establishment, but this is not necessary. Any corrective action is left to the establishment to take voluntarily.
- OAI–Official Action Indicated: Objectionable conditions were found and regulatory action should be recommended. Includes inspections resulting in recalls where the district has decided conditions warrant regulatory action.
You can see the spectrum of potential classifications for each 483 citation. Typically, a classification of OAI results in a warning letter sent to the company and exhibited in all its glory on the FDA website. For a foreign company (outside of the USA) their products may also be placed on import alert; oh, joy of joys. You can now see that is better to be compliant than not. Furthermore, the cost of compliance (the cost of doing it right) is always cheaper than the cost of non-compliance (the cost of getting caught) (5).
Returning to the reason for the new version of CPG 7346.832 above, you can see that one of the reasons for updating the document is to add instructions for potential official action indicated (pOAI) reporting responsibilities (3). These are instructions to inspectors on how to report serious non-compliances that could result in a warning letter to the organization.
Why Does this Document Matter?
You may ask why we should discuss this document now, given that it doesn’t become fully effective until September 2022. A good point. However, consider that any analytical development work conducted that is part of a regulatory submission to the FDA comes under this new version of CPG 7346.832 (3), hence such work may be inspected against these more detailed requirements.
The structure of CPG 7346.832 is shown in Figure 1, and discussed in the following sections of this column. Please note that while in Figure 1 there are areas of reformatting and enhancement, much of the 2010 wording has been retained in the new version. This column looks specifically at the high-level impact on a regulated laboratory, but to gain a full understanding of all of the changes, you will need to read and compare both versions of this document.


Figure 1: Structure of the 2019 version of CPG 7346.832 for preapproval inspections.
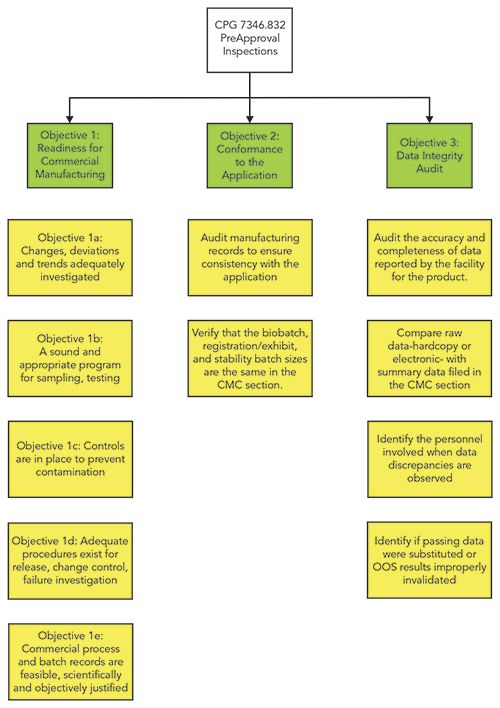
Let us look in more detail at each of the objectives in the new CPG document. Remember that I mentioned that the old version has three objectives with data integrity running through them? Now is your opportunity to see and understand the impact of this new version of the document.
Objective 1: Readiness for Commercial Manufacturing
Objective 1 in the new version simply states:
Determine whether the establishment has a quality system that is designed to achieve sufficient control over the facility and commercial manufacturing operations (3).
You may be thinking from the title that this objective only applies to the manufacturing side, and analytical scientists can relax and put their feet up. Sorry, the new version of CPG 7346.832 retains the five sub-objectives from the old version that focus on the overall Pharmaceutical Quality System (PQS). All of these sub-objectives have impacts on a regulated laboratory, from cleaning validation through sampling to release testing (3):
- Objective 1a: Manufacturing and laboratory changes, deviations, and trends relating to the development of drug substance and drug product manufacturing have been adequately evaluated.
There is an inspection focus on the changes, deviations, and trends within the laboratory on the PAI menu. While this is a feature of all inspections now, it is the changes around a new product that will be subject to scrutiny.
- Objective 1b: A sound and appropriate program for sampling, testing, and evaluating components (including active pharmaceutical ingredients [APIs]), in-process materials, finished products, containers, and closures for purposes of releasing materials or products has been established, including a robust supplier qualification program.
The laboratory does not escape from this sub-objective; you are in it up to your collective necks. If you outsource any analytical work, then how did you select the facility, how much due diligence was performed, and what do the quality and technical agreements cover? A good overview of the requirements for outsourcing is EU GMP Chapter 7 on Outsourcing (6).
Objective 1c: Sufficient facility and equipment controls are in place to prevent contamination of and by the application product (or API).
If your laboratory conducts cleaning validation, guess what? This one’s for you.
Objective 1d: Adequate procedures exist for batch release, change control and investigating failures, deviations, complaints, and adverse events, and for reporting this information to FDA.
How are batches released, and what is the role of laboratory data in this process? Here, the inspector asks whether the procedures for various investigations are adequate rather than investigating deviations for the product itself, which is covered in Objective 1a. Obviously, there will not be any complaints or adverse events, as the product has not been licensed yet; the inspection will focus on determining if the procedures are adequate and meet applicable regulations.
Objective 1e: The proposed commercial process and manufacturing batch record, including instructions, processing parameters, and process control measures, are feasible and scientifically and objectively justified. This objective is linked to the firm’s process validation program across the product lifecycle.
Do your analytical procedures have sufficient detail? Do you collect all records in an analysis? How do you process your analytical data to generate the reportable result? Where are the analytical records stored? What data integrity and good documentation practices are there, and how are staff trained?
Objective 1 in total is pervasive throughout both the organization and specifically any laboratories involved in method development, validation of analytical procedures, sampling, testing, product release, supplier qualification (contract analytical laboratories), change control, deviations and investigations. And by the way, everything the laboratory does must be scientifically sound, including any investigations for invalidating out of specification (OOS) results.
In short:
- Are you in control?
- Are processes formally proceduralized?
- Where is the documented evidence that you follow these procedures?
Objective 2: Conformanceto the Application
The second objective is stated as:
Verify that the formulation, manufacturing or processing methods; analytical (or examination) methods; and batch records are consistent with descriptions contained in the CMC section of the application. This may include CMC information relevant to exhibit batches, biobatches, other pivotal clinical batches, and the proposed commercial-scale process (3).
Unlike the first objective, with a neat and structured list of sub-objectives, the areas for inspection under this objective are presented like a shopping list. Table I lists the pertinent inspection areas for a regulated laboratory together with the inspection focus within each area. Please note that Table I is an edited list; please read the actual document for the full scope of Objective 2. Note that there is a recurring theme in this objective of checks from laboratory data on-site back to the application submitted by the organization.



Some of you may be confused by the term biobatch in Table I; this refers to the manufacturing batch used to make supplies for pivotal clinical studies. It can also be called a pilot batch and is different from commercial batches as the amount synthesized is smaller, and typically there is no predetermined shelf life; rather, the product stability is determined as an ongoing process. As such, it is an inviting inspection target, especially for data integrity.
Analytical procedures that will be high on a list of targets in a PAI will be those that are unique to the product under inspection rather than pharmacopoeial methods. In particular, have any procedures been modified since the application was submitted?
Factual and Contextual Data Integrity
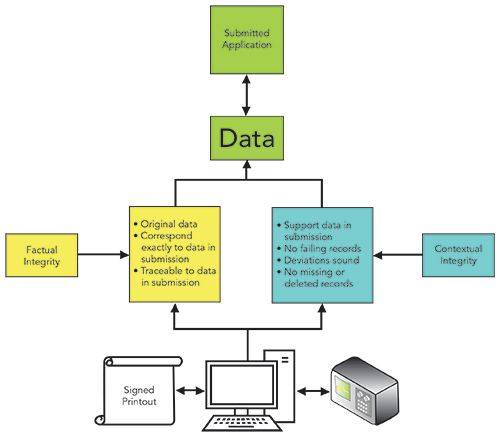
One of interesting asides in the new CPG is the concept of factual and contextual data integrity discussed in a footnote at the end of Objective 2. In the context of a PAI, these terms are defined as:
- Information that has factual integrity is original and corresponds directly to that submitted to FDA (for example, a chromatogram showing a peak area that directly calculates to an assay value submitted in a data summary sheet in the application) (3).
- Information that has contextual integrity supports submitted information about the testing or manufacturing area and related products or processes (for example, a chromatographic sequence that shows all the assayed samples and that does not reveal failing assay values).
- Missing records (batch or testing) and unexplained losses of inventory of components used in production may call into question the contextual integrity of the information filed in an application (3).
Figure 2 shows a hybrid spectrometer system at the bottom that has generated data and records that have been included in an application to the FDA at the top of the page. The journey from the instrument to the application takes two routes; one for factual integrity, and the other for contextual integrity. Factual integrity ensures a match between data and information in the application and the electronic records and associated paper printouts (for hybrid systems) in the data systems of the organizations’s laboratory. In contrast, contextual integrity is an assessment that the organization has not been selective or fraudulent in including data in an application. This concept of factual and contextual integrity provides a bridge between Objectives 2 and 3, as data integrity issues identified in the former provide an input to the latter.


Figure 2: Factual and contextual data integrity.
Objective 3: Data Integrity Audit
The aim of objective 3 is:
Audit and verify raw data at the facility that are associated with the product. This information can, among other things, help to authenticate the data submitted in the CMC section of the application as relevant, accurate, complete, and reliable for CDER assessment (3).
Similar to Objective 2 and dissimilar to Objective 1, there is a shopping list of items that have been extracted, edited, and ordered into topic areas below and as shown in Figure 1.
Audit the accuracy and completeness of data reported by the facility for the product (3):
- Select key data sets from drug development or randomly select data from the application. Generally, data on finished product stability, dissolution, content uniformity, and API impurity are good candidates for this audit.
- Compare raw data-hardcopy or electronic-such as chromatograms, spectrograms, laboratory analyst notebooks, and additional information from the laboratory with summary data filed in application. Raw data files should support a conclusion that the data/information reported by the site are complete and accurate.
- When data discrepancies are observed, identify the firm personnel involved. Determine which actions or inactions contributed to the data integrity problem, and whether corrective actions were or are to be taken.
- Also determine whether data that should have been reported in the application were not reported.
The new version of the CPG shows some indicators of possible laboratory data integrity problems and I have collated them in Table II. The aim of Objective 3 is to demonstrate the reliability of the data in a submission. However, if data are unreliable, the new CPG version suggests that an inspector can expanding the coverage of the inspection to marketed products under Compliance Program 7356.002 for Drug Manufacturing Inspections (7), or that the FDA invokes the Application Integrity Policy (8). If either of these occur, the laboratory has begun the long and very uncomfortable decent into inspection hell.



Working in Research and Development?
Reading this, you can see that the majority of data generated for a drug submission will come from the research and development (R&D) departments. Not all R&D records may be subject to GMP, which can give some R&D departments an air of the wild west, which only tends to come under control when QA comes calling. However, the tendency of R&D to be outside of GMP needs a note of caution as any records submitted to the FDA in Drug Master Files and New Drug Applications are subject to 21 CFR 11 regulations as a stated in 21 CFR 11.1(b) (9):
This part applies to records in electronic form that are created, modified, maintained, archived, retrieved, or transmitted, under any records requirements set forth in agency regulations. This part also applies to electronic records submitted to the agency under requirements of the Federal Food, Drug, and Cosmetic Act and the Public Health Service Act, even if such records are not specifically identified in agency regulations.
R&D data included in FDA drug applications must comply with Part 11 requirements regardless, and GMP regulations where applicable. The validation of analytical procedures, any testing of components, APIs, and so forth must have the same regulatory approaches as QC.
However, there are also key business reasons why R&D and QC should have the same approaches to instrument qualification, computerized system validation and validation of analytical procedures and testing:
- no dual standards
- ability to move QC staff into R&D and vice versa
- more efficient technology transfer.
You know it makes sense….. but you’ll probably ignore this advice.
Summary
The new version of CPG 7346.832 for Pre-Approval Inspections is effective now but is fully effective by September 2022. It is better to understand the implications and implement any changes now to ensure that any work subject to a PAI meets these updated requirements. Even if you are not involved in developing or validating analytical procedures or testing for regulatory submissions, the new version of CPG 7346.832 provides you with an insight into the way that an FDA inspector will conduct an inspection. For readers working in Quality Assurance roles, it also gives you ways of conducting data integrity audits.
References
- Able Laboratories Form 483 Observations. 2005 1 Jan 2016]; Available from: http://www.fda.gov/downloads/aboutfda/centersoffices/officeofglobalregulatoryoperationsandpolicy/ora/oraelectronicreadingroom/ucm061818.pdf.
- FDA Compliance Program Guide CPG 7346.832 Pre-Approval Inspections (Food and Drug Adminsitration, Silver Spring, Maryland, 2010).
- FDA Compliance Program Guide CPG 7346.832 Pre-Approval Inspections (Food and Drug Adminsitration, Silver Spring, Maryland, 2019).
- Field Management Directive (FMD) 86: Establishment Inspection Report Conclusions and Decisions (Food and Drug Adminsitration, Silver Spring, Maryland, 2014).
- R.D.McDowall, Data Integrity and Data Governance: Practical Implementation in Regulated Laboratories (Royal Society of Chemistry, London, United Kingdom, 2019).
- EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 7 Outsourced Activities (European Commission: Brussels, Belgium, 2013).
- FDA Compliance Program 7456.002 Drug Manufacturing Inspections (Food and Drug Adminsitration, Silver Spring, Maryland, 2017).
- FDA Application Integrity Policy: Fraud, Untrue Statements of Material Facts, Bribery, and Illegal Gratuities (Compliance Policy Guide Section 120.100) (Food and Drug Adminsitration, Silver Spring, Maryland, 1991).
- 21 CFR 11 Electronic records; electronic signatures, final rule, in Title 21 (Food and Drug Administration, Washington, DC, 1997).



R.D. McDowall is the director of R.D. McDowall Limited and the editor of the “Questions of Quality” column for LCGC Europe, Spectroscopy’s sister magazine. Direct correspondence to: SpectroscopyEdit@MMHGroup.com














