Improving Oligonucleotide Sensitivity and Separation for LC-MS Applications
The authors discuss the use of reversed-phase LC-MS for oligonucleotide characterization.
Ion-pairing assisted reversed-phase high performance liquid chromatography (HPLC) has been a common method for purifying oligonucleotides for some time, but has not been widely used for routine quality control methods that assess purity. Ion-exchange HPLC, matrix-assisted laser desorption–ionization mass spectrometry (MALDI-MS), and capillary electrophoresis have been used more commonly for determining oligonucleotide purity. With the development of several potential human therapeutic oligonucleotide drugs, many manufacturers are now looking to use LC–MS as an identity test as well as to quantitate and identify synthetic impurities.
Oligonucleotides are organic polymers in which nucleosides (an aromatic base attached to a ribose sugar) are linked by a phosphodiester into long chains up to thousands of bases in length.
The combination of aromatic bases, polar sugars, and polyanionic phosphates can make separations of oligonucleotides very challenging; many will only differ slightly in their overall charge and hydrophobicity based upon their base composition and length. Oligonucleotides from natural sources are typically thousands of bases in length and are more amenable to separation using molecular biology techniques. Oligonucleotide therapeutics and oligonucleotide reagents are much shorter in length (from 2 to 100 bases in length) and usually are generated through synthetic methods using stepwise chemical synthesis. Such synthetic oligonucleotides usually are purified and analyzed using chromatographic techniques; often ion-exchange chromatography is used to purify the desired full-length oligonucleotides from shorter length synthesis impurities. Reversed-phase chromatography also is used for purifying synthetic oligonucleotides; however, their high polarity requires the use of an ion-pairing buffer (typically triethylamine or other alkyl amines) to increase retention and improve selectivity (1).
Interest in quality control analytical methods for oligonucleotides has grown in the last few years as oligonucleotide therapeutic candidates have been passing through the research phase of product development and into clinical trials (2). Traditionally, ion-exchange high performance liquid chromatography (HPLC), capillary electrophoresis, polyacrylamide gel electrophoresis (PAGE), and matrix-assisted laser desorption–ionization (MALDI)-MS have been used to characterize oligonucleotides; however, each method has unique difficulties in quantitating and characterizing minor impurities. Lately, manufacturers have looked at adopting reversed-phase LC–MS for characterizing impurities in oligonucleotide products (2). Reversed-phase LC–MS has unique advantages in that impurities can be separated chromatographically from a parent and then identified and quantitated based upon MS or tandem MS-MS analysis. However, using reversed-phase LC–MS is not without its limitations. Modifications to the ion-pairing mobile phase to allow for MS compatibility often result in reduced resolution of oligonucleotides compared to traditional ion-pairing reversed-phase HPLC separation methods. Sensitivity also can be an issue in that ion-pairing reagents are known suppressors of electrospray MS; however, such reagents are needed when working with oligonucleotides to achieve retention on reversed-phase columns (3,4).
Research was undertaken to improve LC–MS sensitivity by investigating different concentrations of ion-pairing buffers to achieve the best MS signal while maintaining adequate selectivity between oligonucleotides that differ by only one residue in length. Different MS systems were used as well to determine if mobile phase conditions were MS-dependent or specific to a particular interface. Because TEA–HFIP buffer mixtures are commonly used for oligo MS work, the ratio between the buffers was varied to determine the mixture that delivered the optimal MS signal while maintaining resolution.
Experimental
Oligonucleotide samples were run on an 1100 HPLC (Agilent Technologies, Wilmington, Delaware) with quaternary pump module, autosampler, and a diode-array detector. The column oven temperature was set at 50 °C and various gradients were used, depending upon the sample and mobile phase used for analysis. LC–MS analysis of oligonucleotides was performed using either a 3000 triple quadrupole, 4000QTrap hybrid-triple quadrupole-LIT (Applied Biosystems, Foster City, California) or the Esquire ion-trap MS system (Bruker, Billerica, Massachusetts). All three MS systems were operated in negative ion mode (because oligonucleotides are polyanionic) and sample was introduced into each MS system using their standard electrospray interfaces with optimized operating conditions. An m/z range from 500 to 2200 was monitored in total ion current (TIC) mode for each instrument. Molecular weight deconvolution–deconstruction was performed using the Bayesian Protein Reconstruct tool within BioAnalyst (Applied Biosystems).
The HPLC column used for all the separations was a 150 mm x 2.0 mm Clarity 3-µm Oligo-RP column (Phenomenex, Inc., Torrance, California), an HPLC medium specifically designed for oligonucleotide separations. Mobile phase was varied run-to-run (see specific figures for composition), but consisted of a combination of TEA and HFIP in 98% water and 2% methanol for the aqueous mobile phase. For the organic mobile phase, TEA and HFIP were mixed in with 98% methanol and 2% water. The gradient was from 5% to 10% organic in 5 min followed by an increase from 10% to 30% organic in 15 min. For the last set of chromatograms, the second gradient was shallower — 10–30% organic in 60 min.


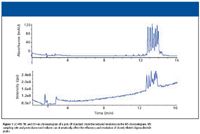
Figure 1
LC–MS Considerations
A separation of an oligonucleotide mixture (poly dT 12-18) separated by reversed-phase HPLC is shown in Figure 1. Data were collected on both UV detector (Figure 1a) and mass spectrometer (Figure 1b) linked in series. Note the reduced separation of the different oligonucleotides in the TIC trace from the MS system. The reduced resolution is related to the additional postcolumn void volume introduced by both the UV detector and the MS interface linked in series. Slow MS sampling rates also can influence the overall resolution and peak width of separated oligonucleotides. While such a loss of resolution might not be of great concern for simple mixtures of oligonucleotides, for complex mixtures of isobaric components, such a loss of resolution can make impurity quantitation difficult. This is complicated further by the fact that most therapeutic oligonucleotides are 18–40-mer in length (around 5–12 kDa molecular weight). Because most electrospray ionization (ESI) mass spectrometers monitor between 300 and 2200 m/z, molecular ions from –5 to –15 charge state are the predominant ions detected in an ESI mass spectrometer. To fully quantitate oligonucleotide species, reconstruction software is required to generate the parent mass. An example is shown in Figure 2, where a 21-mer DNA was analyzed by LC–MS using a shortened gradient. Figure 2a is the TIC of the LC–MS run, with the raw spectra for the principal peak shown in Figure 2b. The reconstructed mass of the oligonucleotide is shown in Figure 2c. The use of reconstruction software further emphasizes the importance of having optimal chromatography for LC–MS of oligonucleotides.



Figure 2
Mobile Phase Optimization
For oligonucleotide purification by reversed-phase HPLC, researchers generally use TEAA buffer in high concentrations (100 mM) for ion-pairing. To adapt MS detection to reversed-phase separation of oligonucleotides, others have shown applications using a combination of TEA and HFIP (generally 15 mM TEA–400 mM HFIP) for LC–MS separations of oligonucleotides (5). This mobile phase combination of ion-pairing reagents TEA and HFIP is more compatible for MS sensitivity; however, there is a concern that such conditions compromise the separation of oligonucleotides and reduce the sensitivity of MS detection due to ion suppression.
Conditions were varied to improve LC–MS sensitivity in reversed-phase oligonucleotide QC methods while maintaining enough ion-pairing reagent to maintain acceptable oligonucleotide selectivity. Poly-dT standards were run at different mobile phase concentrations to determine which conditions gave the best selectivity and resolution. HFIP–TEA concentrations were varied from 50 mM HFIP–2 mM TEA to 400 mM HFIP–16 mM TEA. An overlay of such separations is shown in Figure 3. The results are interesting in that mobile phase conditions using low amounts of ion-pairing buffer (50 mM HFIP–2 mM TEA) gave reduced MS signal and oligo retention (as well as selectivity). This is contrary to the hypothesis that reduced ion-pairing reagent delivers improved MS sensitivity, but is easily explained due to the reduced retention of the oligonucleotide, resulting in an elution in a lower percentage of organic mobile phase. It has been widely observed that increasing organic content in an electrospray interface results in improved MS sensitivity; such an observed decrease in MS signal using low amounts of ion-pairing reagent is consistent with this observation. Further investigation also reveals a reduction in MS signal intensity for higher levels of ion-pairing reagent despite an increase in retention. Such observations match with expected results and suggest that ion suppression is occurring at high levels of ion-pairing reagent. These initial results suggest that a mobile phase containing 4–8 mM TEA and 100–300 mM HFIP is required for optimal separation and signal for this application.



Figure 3
Additional studies were performed to determine if different ratios between TEA and HFIP provide differences in resolution and MS sensitivity. The previous experiment suggests that obtaining maximum resolution and MS sensitivity revolves around maintaining good retention of the ion-paired oligonucleotide. Different ratios were investigated based upon using a fixed amount of HFIP (200 mM) and TEA concentrations ranging from 1 mM to 14 mM. Results are shown in Figure 4. The pH of the resulting mobile phase was tested and is shown in the figure. Findings suggest that the ratio between HFIP and TEA is critical in obtaining the maximum retention, MS sensitivity, and selectivity. Similar to what was observed in Figure 3, results suggest that the optimal mobile phase for maximum MS sensitivity and oligonucleotide separation is a balance between obtaining maximum retention (via ion-pairing reagent concentration) and minimizing MS suppression. It appears that mobile phase conditions using 200 mM HFIP and 8 mM TEA (pH 8.0) provided the best results for LC–MS analysis for this mixture. Separating a different sample or using a different HPLC column likely will require different optimal conditions.



Figure 4
Conclusions
Unlike other techniques for oligonucleotide characterization, reversed-phase LC–MS offers the capability to positively identify the analytes in an oligonucleotide mixture. Reducing extracolumn volume by removing the UV detector from the flow path as well as optimizing MS conditions can improve oligonucleotide LC–MS resolution significantly. Improved resolution can decrease the complexity of the MS spectra, making the results generated from deconvolution software more useful.
Decreasing the amount of ion-pairing reagent can improve MS signal for oligonucleotides, but only to a point. Loss of retention under conditions with low ion-pairing reagent concentration actually can reduce MS signal (due to earlier elution in lower organic mobile phase). Reduced ion-pairing reagent also reduces resolution of closely eluted oligonucleotides. For this study, the optimal mobile phase conditions for obtaining good resolution and MS signal were found to be 200 mM HFIP–8 mM TEA. While not tested here, different types of oligonucleotides (RNA or phosphorothioate DNA) likely will have different optimum conditions based upon their different hydrophobicities.
Greg Scott and Michael McGinley are with Phenomenex, Inc., Torrance, California. Matthew Champion is with Applied Biosystems, Foster City, California.
References
(1) R. Deshmukh, M. Leitch, Y. Sanghui, and D. Cole, in Handbook of Bioseparations, S. Ahuja, Ed. (Academic Press, New York, 2000), pp. 511–534.
(2) G. Hannon and J. Rossi, Nature 431, 371–378 (2004).
(3) C. Castleburry, L. Sallans, R. Kepler, and P. Limbach, Oral Presentation at Pittcon 2008, "LC/MS/MS analysis of phosphorothioate oligonucleotides."
(4) M. Hall, B. Elliott, and K. Anderson, American Biotechnol. Lab. Appl. Supp., January 2004.
(5) M. Gilar, K. Fountain,Y. Bulman, J. Holyoke, H. Dovoudi, and J. Gebler, Oligonucleotides 13, 229–243 (2003).














High-Speed Laser MS for Precise, Prep-Free Environmental Particle Tracking
April 21st 2025Scientists at Oak Ridge National Laboratory have demonstrated that a fast, laser-based mass spectrometry method—LA-ICP-TOF-MS—can accurately detect and identify airborne environmental particles, including toxic metal particles like ruthenium, without the need for complex sample preparation. The work offers a breakthrough in rapid, high-resolution analysis of environmental pollutants.


The Fundamental Role of Advanced Hyphenated Techniques in Lithium-Ion Battery Research
December 4th 2024Spectroscopy spoke with Uwe Karst, a full professor at the University of Münster in the Institute of Inorganic and Analytical Chemistry, to discuss his research on hyphenated analytical techniques in battery research.

Mass Spectrometry for Forensic Analysis: An Interview with Glen Jackson
November 27th 2024As part of “The Future of Forensic Analysis” content series, Spectroscopy sat down with Glen P. Jackson of West Virginia University to talk about the historical development of mass spectrometry in forensic analysis.

Detecting Cancer Biomarkers in Canines: An Interview with Landulfo Silveira Jr.
November 5th 2024Spectroscopy sat down with Landulfo Silveira Jr. of Universidade Anhembi Morumbi-UAM and Center for Innovation, Technology and Education-CITÉ (São Paulo, Brazil) to talk about his team’s latest research using Raman spectroscopy to detect biomarkers of cancer in canine sera.


