Limitations in Analytical Accuracy, Part I: Horwitz's Trumpet
Spectroscopy
September 2006. In the first part of this two-part series, columnists Jerome Workman, Jr. and Howard Mark discuss the limitations of analytical accuracy and uncertainty.



Two technical papers recognized as significant early contributions in the discussion of the limitations of analytical accuracy and uncertainty include those by Horwitz of the U.S. FDA (1,2). For this next series of articles, we will be discussing both the topic and the approaches to this topic taken by the classic papers just referenced. The determination and understanding of analytical error is often approached using interlaboratory collaborative studies. We have previously delved into that subject in "Chemometrics in Spectroscopy" with a multipart column series (3–8).



Jerome Workman, Jr.
Horwitz points out in his Analytical Chemistry A-pages paper (1), inserting the statement made by John Mandel, "the basic objective of conducting interlaboratory tests is not to detect the known statistically significant differences among laboratories: 'The real aim is to achieve the practical interchangeability of test results.' Interlaboratory tests are conducted to determine how much allowance must be made for variability among laboratories in order to make the values interchangeable."



Howard Mark
Horwitz also points out the universal recognition of irreproducible differences in supposedly identical method results between laboratories. It has even been determined that when the same analyst is moved between laboratories, the variability of results obtained by that analyst increases. One government laboratory study concluded that variability in results could be minimized only if one was to "conduct all analyses in a single laboratory . . . by the same analyst." So if we must always have interlaboratory variability, how much allowance in results should be regarded as valid — or legally permissible — as indicating "identical" results? What are the practical limits of acceptable variability between methods of analysis, especially for regulatory purposes?
We will address aspects of reproducibility, which has been defined previously as "the precision between laboratories." It also has been defined as "total between-laboratory precision." This is a measure of the ability of different laboratories to evaluate each other. Reproducibility includes all the measurement errors or variances, including the within-laboratory error. Other terms include precision, defined as "the closeness of agreement between independent test results obtained under stipulated conditions" (9); and repeatability, or "the precision for the same analyst within the same laboratory, or within-laboratory precision." Note that for none of these definitions do we require the "true value for an analytical sample." In practice, we do not know the true analyte value unless we have created the sample, and then it is only known to a given certainty (that is, within a determined uncertainty).
Systematic error is also known as bias. The bias is the constant value difference between a measured value (or set of values) and a consensus value (or true value if known). Specificity is the analytical property of a method or technique to be insensitive to interferences and to yield a signal relative to the analyte of interest only. Limit of reliable measurement predates the use of minimum detection limit (MDL). The MDL is the minimum amount of analyte present that can be detected with known certainty. Standard error of the laboratory (SEL) represents the precision of a laboratory method. A statistical definition is given in the following paragraph. The SEL can be determined by using one or more samples properly aliquoted and analyzed in replicate by one or more laboratories. The average analytical value for the replicates on a single sample is determined as



SEL is given by


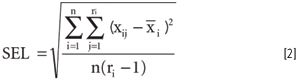
where the i index represents different samples and the j index different measurements on the same sample.



Any analytical method inherently carries with it limitations in terms of speed, allowable uncertainty (as MDL), and specificity. These characteristics of a method (or analytical technique) determine where and how the method can be used. Table I shows a template to relate purpose of analytical methods to the speed of analysis and error types permitted.


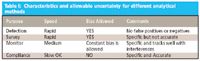
Table I: Characteristics and allowable uncertainty for different analytical methods
Bias is allowed between laboratories when constant and deterministic. For any method optimization, we must consider the requirements for precision and bias, specificity, and MDL.
Horwitz claims that irrespective of the complexity found within various analytical methods, the limits of analytical variability can be expressed or summarized by "plotting the calculated mean coefficient of variation (CV), expressed as powers of two [ordinate], against the analyte level measured, expressed as powers of 10 [abscissa]." In an analysis of 150 independent Association of Official Analytical Chemists (AOAC) interlaboratory collaborative studies covering numerous methods such as chromatography, atomic absorption, molecular absorption spectroscopy, spectrophotometry, and bioassay, it appears that the relationship describing the CV of an analytical method and the absolute analyte concentration is independent of the analyte type or the method used for detection.
Moving ahead to describe the details of this claim, we need to develop a few basic review concepts. The standard deviation of measurements is determined by first calculating the mean, then taking the difference of each control result from the mean, squaring that difference, dividing by n – 1, then taking the square root. All of these operations are implied in the following equation:

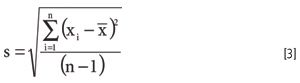



The data for Figures 1 and 2 are shown in Tables II and III, respectively.


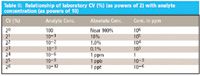
Table II: Relationship of laboratory CV (%) (as powers of 2) with analyte concentration (as powers of 10)
In reviewing the data from the 150 studies, it was found that about 7% of all data reported could be considered outlier data as indicated by a Dixon test. Some international refereed methods performed by experts had to accept up to 10% outliers resulting from best efforts in their analytical laboratories.


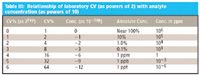
Table III: Relationship of laboratory CV (as powers of 2) with analyte concentration (as powers of 10)
Horwitz throws down the gauntlet to analytical scientists, stating that a general equation can be formulated for the representation of analytical precision. He states this as follows:
CV(%) = 2(1–0.5log C)
where C is the mass fraction of analyte as concentration expressed in powers of 10 (for example, 0.1% is equal to C = 10–3).


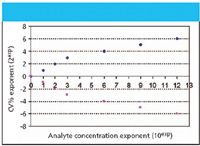
Figure 1: Relationship of laboratory CV (as %) with analyte concentration as powers of 10exp. (For example, 6 on the abscissa represents a concentration of 106, or 1 ppm.) Note: The shape of the curves has been referred to as Horwitz's trumpet.
At high (macro) concentrations, CV doubles for every order of magnitude that concentration decreases; for low (micro) concentrations, CV doubles for every three orders of magnitude decrease in concentration. Note that this represents the between-laboratory variation. The within-laboratory variation should be 50–66% of the between-laboratory variation. Reflecting on Figures 1 and 2, some have called this Horwitz's trumpet. How interesting that he plays such a tune for analytical scientists.


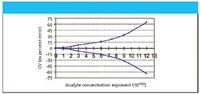
Figure 2: Relationship of laboratory CV (as powers of 2) with analyte concentration as powers of 10âexp. (For example, 6 on the abscissa represents a concentration of 1010â6, or 1 ppm with a CV (%) of 24.)
Another form of expression also can be derived, because CV (%) is another term for percent relative standard deviation (% RSD) as follows (12):
%RSD = 2(1–0.5log C)
There are many tests for uncertainty in analytical results and we will continue to present and discuss these within this series.
Erratum
A sharp-eyed (and careful) reader found a typographical error in our previous column (1). The "A" on the left-hand side of equation 12b should have the subscript 2, rather than 1. Our thanks to Kaho Kwok for pointing this out to us.
(1) Spectroscopy 21(5), 34–38 (2006).
Jerome Workman, Jr. serves on the Editorial Advisory Board of Spectroscopy and is director of research, technology, and applications development for the Molecular Spectroscopy & Microanalysis division of Thermo Electron Corp. He can be reached by e-mail at: jerry.workman@thermo.com
Howard Mark serves on the Editorial Advisory Board of Spectroscopy and runs a consulting service, Mark Electronics (Suffern, NY). He can be reached via e-mail: hlmark@prodigy.net
References
(1) W. Horwitz, Anal. Chem. 54(1), 67A–76A (1982).
(2) W. Horwitz, R.K. Laverne, W.K. Boyer, J. Assoc. Off. Anal. Chem. 63(6), 1344 (1980).
(3) J. Workman and H. Mark, Spectroscopy 15(1), 16–25 (2000).
(4) J. Workman and H. Mark, Spectroscopy 15(2), 28–29 (2000).
(5) J. Workman and H. Mark, Spectroscopy 15(5), 28–29 (2000).
(6) J. Workman and H. Mark, Spectroscopy 15(6), 26–27 (2000).
(7) J. Workman and H. Mark, Spectroscopy 15(7), 16–19 (2000).
(8) J. Workman and H. Mark, Spectroscopy 15(9), 27 (2000).
(9) ASTM E177 – 86. Form and Style for ASTM Standards, ASTM International, West Conshohocken, PA. ASTM E177 – 86 "Standard Practice for Use of the Terms Precision and Bias in ASTM Test Methods."
(10) S. Helland, Scand. J. Statist. 17, 97 (1990).
(11) H. Mark and J. Workman. Statistics in Spectroscopy (second edition) (Elsevier, Amsterdam, 2003), pp. 205–211; 213–222.
(12) Personal communication with G. Clark Dehne, Capital University (Columbus, Ohio) (2004).








From Classical Regression to AI and Beyond: The Chronicles of Calibration in Spectroscopy: Part I
February 14th 2025This “Chemometrics in Spectroscopy” column traces the historical and technical development of these methods, emphasizing their application in calibrating spectrophotometers for predicting measured sample chemical or physical properties—particularly in near-infrared (NIR), infrared (IR), Raman, and atomic spectroscopy—and explores how AI and deep learning are reshaping the spectroscopic landscape.






