New Approaches in Sample Preparation and Precise Multielement Analysis of Crude Oils and Refined Petroleum Products Using Single-Reaction-Chamber Microwave Digestion and Triple-Quadrupole ICP-MS
Spectroscopy
October’s AP column highlights a team of geochemists at the University of Houston who have been developing methods to streamline multi-element analysis for a more complete fingerprinting of oils by using one sample preparation method utilizing a single reaction chamber microwave digestion system and then analyzing these solutions for major, and minor elements by ICP-OES and low abundance trace elements by triple quadrupole (QQQ) ICP-MS. Results to date using this approach have shown that complete elemental recovery and removal of organic matrices can be achieved safely and that up to 57 elements can be determined in oils with good accuracy and precision. Removal of organic matrices during digestion not only helps to limit the formation of polyatomic spectral interferences, but improves instrument stability and reduces carbon build in the sample introduction and interface regions, which have traditionally plagued “dilute and shoot” methods.
This month’s “Atomic Perspectives” highlights a team of geochemists at the University of Houston that has been developing methods to streamline multielement analysis for a more complete fingerprinting of oils by using a single-reaction-chamber microwave digestion sample preparation method and then analyzing for major and minor elements by inductively coupled plasma–optical emission spectrometry (ICP-OES) and low-abundance trace elements by inductively coupled plasma–mass spectrometry (ICP-MS) with a triple-quadrupole MS system. Results to date using this approach have shown that complete elemental recovery and removal of organic matrices can be achieved safely and that up to 57 elements can be determined in oils with good accuracy and precision. The removal of organic matrices during digestion not only helps to limit the formation of polyatomic spectral interferences, but improves instrument stability and reduces carbon buildup in the sample introduction and interface regions, which has traditionally plagued “dilute-and-shoot” methods.
Comprehensive multielement determinations in crude oil and refined petroleum products have been a difficult task, historically resulting in myriad developed sample preparation and instrumental analytical methods (1). These methods are somewhat limited and many do not include all elements that are important to the petroleum industry. Most methods previously developed tend to have limited numbers of targeted elements, which are often determined based on specific method detection limits (MDL), method quantification limits (MQL), or a specific single element need. The quality and comprehensive nature of a suite of elements measured can therefore be dependent on a number of factors:
- suitability of sample preparation techniques and resultant element recovery for specific analytes;
- sample matrix, the weight of the sample prepared, and the necessary dilution factors;
- sensitivity of the analytical instruments to be used;
- nature and availability of certain nonstandard instrument sample introduction systems;
- the ability to mitigate complex spectral interferences, especially for routine analysis of many low-abundance elements;
- quality of standards available;
- purity of the reagent and blanks, impacting the accuracy and precision; and
- repeatability of the method for particular analytes.
As a result of these limitations, the elemental suite is typically limited and also different from one method to the next.
Source of Elemental Constituents in Crude Oil and Petroleum Products
Along with their dominant organic major elemental constituents (for example, C, H, O, and N), crude oil and refined petroleum products are highly variable in minor to ultratrace elemental abundances. Trace and minor elements are generally complexed in the heavier organic molecules or inorganic salts and typically constitute less than 1% by weight of the sample. In general, organic matrices pose major obstacles for a routine method of comprehensive multielement analysis when trying to adopt a single sample preparation technique. A single analytical instrument would require an exceptionally wide dynamic range necessary to cover broad inorganic elemental concentrations that can range from 5% to 6% (50–60 mg/g), such as sulfur, down to minor elements at ~1000–2000 µg/g, such as vanadium, and to ultratrace element concentrations of <1 µg/g, such as the rare earth elements. For this reason, a single crude oil multielement analysis method is elusive without multiple, often complex and time consuming sample preparation or elemental group separation steps. In fact, most elements detectable in crude oils are low in abundance (nanograms per gram or less) and many are plagued by interferences when using traditional single-quadrupole-based inductively coupled plasma–mass spectrometry (ICP-MS) instrumentation. Thus, comprehensive analyses of trace elements in organic matrices by either inductively coupled plasma–optical emission spectrometry (ICP-OES) or ICP-MS have proven to be a challenging problem. Whereas ICP-MS technology has the added sensitivity and multielement capabilities desired for low abundances, there are many hurdles to overcome using a single sample preparation technique.
In this study, we describe a sample preparation method that uses a microwave digestion system with a single reaction chamber (UltraWAVE, Milestone Inc.). This method is capable of producing an organic-free sample solution (<0.1% total carbon) used for analysis of up to 55–57 elements with the appropriate ICP technique (2,3). In this approach, we use in tandem an ICP-OES system (Model 725, Agilent Technologies) for quantitation and screening of higher abundance elements and an ICP-MS system with a triple-quadrupole mass spectrometer (Model 8800, Agilent Technologies) specifically to allow interference-free analysis of low-abundance elements. The single digestion procedure with these two instrumental techniques accommodates the wide range of elemental concentrations necessary for characterizing crude and refined oils in a more comprehensive and routine manner.
Oil Sample Preparation Methods
ICP-MS is now the standard technique for low-abundance elemental analysis because of its high sensitivity for measurements at parts per billion (nanograms per gram) or lower levels. Sample preparation generally contributes the most to the inaccuracy, poor precision, or lack of reproducibility through either incomplete dissolution, contamination, or analyte loss during the dissolution or because of solubility issues. Safe, clean, and full-recovery sample preparation of combustible crude oils for multielement analysis is difficult because of the high organic matrix, element volatility, solution solubility, possible contaminants with various methods, and, to a certain extent, sample sizes that may have to be constrained by certain closed system preparation vessels because of safety or sample loss issues. Four approaches to sample preparation for elemental analysis have generally been used for crude oils and their refined petroleum products (1,4–6):
- Strong acid “wet” digestions by open vessel, closed high pressure–temperature bomb or closed high pressure–temperature microwave-assisted mineralization
- Mineralization by high-pressure combustion in a chamber or ashing followed by strong acid digestions
- Direct sample introduction by dilution with organic solvents
- Direct sample introduction of an emulsion or microemulsion
The latter two have tended to be judged as safer, more expedient, and simpler for the analyst. A drawback for solvent dilution is that it tends to result in high dilution factors (100–1000-fold for heavier oils) and can result in heavy organic loads for the instruments used. They have proven successful in some limited analyte applications, but also have been considered a safer decomposition than high pressure and temperature used in mineralization procedures with bombs or closed-vessel rotor-based microwave systems. They remain somewhat limited in analysis of the large range of elements that can be detected by some methods in crude oils or fuel oils. In all of these methods, clean facilities, ultrapure or doubly distilled reagents including ultrapure water, clean, and low contaminant sample vessels, and well-developed cleaning protocols are critical for high precision and accuracy as well as better detection capability.
The “Dilute-and-Shoot” Approach
As stated above, commonly used methods of direct sample introduction known as the dilute-and-shootmethods involve dilution and stabilization of the hydrocarbon sample with an organic solvent, such as toluene, xylene, or kerosene, that is aspirated directly into an argon plasma for routine quantification by ICP-OES, with more recent attempts to adapt the technique for ICP-MS (1,6). In these solvent methods, the instruments have to be standardized with organic solutions with trace-element compounds that are also soluble in organic solvents. There are, however, a few natural crude or fuel oil standards (for example, National Institute of Standards and Technology [NIST] standard reference materials [SRMs]) and generally they are certified for a small number of elements (such as V, Co, Ni, Hg, or S) with additional noncertified reference or information values reported for some elements within some standards. In addition, there are multiple analytical methods used because of the difficulties in certification or the referencing process. For example, for the NIST 1634c residual fuel oil examined here, seven different analytical methods were used for three certified elements, two noncertified referenced elements, and three noncertified information values (seven methods, eight elements to achieve reported values). Such a suite of instruments and analytical techniques would be impractical for routine multielement analysis and extremely time consuming. Synthetic organometallic standards can be purchased with more elements certified, but often they are expensive and tend to have limited stability and shelf life. They are useful in high-throughput laboratories where long shelf stability is typically not required, but may cause analysis repeatability issues in some laboratories. Another drawback of these direct ICP-OES introduction methods is the lower sensitivity of the ICP-OES compared to the more sensitive ICP-MS. This and the high method dilution factors will limit the number of elements that can be successfully quantified. This limitation has led to method developments that analyze a greater numbers of elements with dilute-and-shoot organic solutions (1) or microemulsions (5) that have been adapted directly to ICP-MS. These direct introduction techniques are not as simple for ICP-MS because of deleterious effects on plasma stability, complex ionization effects, deposition or clogging of carbon on the sampler or skimmer cones, carbon-based interferences caused by the solvent, much longer wash out times, and crude oil solubility issues. Some of these effects can be mitigated by desolvation or ultrasonic nebulization techniques (1). Preparation by emulsification with water offers some of the same problems in terms of larger dilution factors and multiple short-term solution analyte stability issues, but it does remove the necessity of organic standards.
Combustion Methods
Our investigations of open- and closed-vessel “combustion” of standard multielement certified oils, followed by strong acid digestions as a method of preparation, showed that both contamination and some very severe elemental losses occur, demonstrating that the combustion method is probably unsuitable for the broad multielement analyses of the large element suites that we are attempting. Although, in part, the dilute-and-shoot solvent, emulsification, and combustion methods as a group have been successfully applied for select groups of targeted elements, they have not resulted routinely in extensive element suites measured using ICP methods for more complete fingerprinting of elemental abundances in crude oil or refined products. Nor have they consistently proven to be high precision or highly repeatable in intra- and interlaboratory studies (1). They are important, however, for some targeted applications and analytical needs.
Acid Digestion Techniques
For strong acid–based digestions that attempt to destroy the organic matrix, closed-vessel digestion procedures are typically advantageous to minimize the analyte loss, but sample sizes have generally been limited to 0.1–0.25 g to avoid damage to the digestion apparatus at high pressures or sample loss and this can limit detection. Alternatively, open-vessel digestion procedures offer the ability to dissolve much larger samples under relatively mild conditions, but are subject to sample loss and contamination. In a recent study by Yasnygina and colleagues (7), the analysis of 39 elements by ICP-MS was attempted using a technique of heating a large sample of 3.5 g of crude oil in strong acid and peroxide in quartz tubes, followed by ashing at 300 °C for 8 h in a low-pressure muffle furnace. Their analytical results based on four replicates of natural oils showed percent relative standard deviations (%RSDs) for 14 elements were better than 10%, but the remaining 25 showed poorer precisions ranging from 10% to 50%. Open vessels can be used, however, after acid digestions to eliminate residual concentrated acids and volatile matrix constituents by evaporation in ultraclean high-efficiency particulate arrestance (HEPA) filtered hood environments. The microwave-assisted strong acid digestion method described here (2,3) combines the use of larger sample sizes and microwave closed system techniques, with an approach using a recently developed single-reaction-chamber microwave system that results not only in good recoveries, but more efficient throughput, larger samples sizes, and enhanced safety.
Microwave-Assisted Strong Acid Wet Digestion Techniques
In general, it is well-accepted that decompositions (mineralization) are more robust and accurate than dilute and shoot methods, but they have been considered more time consuming and to date have not produced more comprehensive data sets with a single methodology. More common rotor-based closed microwave systems have been utilized in the past to prepare crude or fuel oil samples, including SRMs. Wondimu and Goessler (8), for example, conducted these digestions on NIST fuel oil 1634c, but their digestions were limited in sample size because testing showed vessel venting and sample loss if sample sizes were greater than ~0.25 g. However, this 0.25-g weight is well below the 1.0-g sample size recommended by NIST based on their sample homogeneity tests. The investigators were still able to successfully measure 17 out of 24 elements on NIST residual fuel oil 1634c with <5% RSD and good recovery using their digests with ICP-MS. Ducyk and colleagues (9) likewise were able to quantify nine elements in crude oil, and up to 15 in trace element-enriched asphaltene fractions of the crude oil by rotor-based microwave digestion and ultrasonic nebulization coupled with ICP-MS. Periera and colleagues (10) compared microwave-assisted digestion for up to 12 elements with a novel microwave-induced combustion method to achieve multielement analysis of up to 17 elements that were above MDL using both ICP-OES and ICP-MS for trace element-enriched heavy oils, although more elements were attempted. These methods were among several that showed the promise of microwave assisted digestions at least for smaller samples but wider suites of elements were still not possible because of the higher MDL and MQL.
At the University of Houston we have carried out exhaustive testing of various sample preparation techniques including
- high pressure combustion followed by acid digestion;
- high pressure–temperature acid digestions;
- microwave assisted high pressure–temperature acid digestions; and
- the use of strong acids and reagent mixtures for digestion, using various other vessel types.
These approaches have all been explored in an attempt to develop methodologies that can produce a more comprehensive elemental suite with a single method. These methods have been previously summarized in an American Chemical Society (ACS) webinar (2), conference proceedings (3), and more-detailed procedural outlines and fully compiled data sets on multiple NIST standards and crude samples will appear in two upcoming papers by Casey and colleagues (under review) and Yang and colleagues (under review) (20,21). We found that combustion methods resulted in low recovery of many of the analyte target elements. We also concluded that whereas mineralization approaches in closed-system high pressure–temperature bombs with PTFE vessels are acceptable for analysis, they are more time consuming and lacked the adequate sample size characteristics necessary for desired sample throughput, sample homogeneity, and the required MDL and MQL. The mineralization procedures in the microwave single-reaction-chamber high pressure–temperature system using high-purity quartz glass vessels and ultrapure strong acids and reagents alternatively offers clear advantages for both mineralization and more complete analysis of extensive suites of up to 57 elements. Exact procedures, methodologies, and more extensive data will be presented in the future papers listed above, but we provide a general overview of the methodology in this study.
Single-Reaction-Chamber Microwave Digestion
Let’s take a closer look at the methodology we have developed in our laboratory with a microwave digestion system using mixtures of concentrated acids such as HNO3, HCl, and 30% H2O2 in high-purity quartz tubes. With an approximately 1.0-g crude oil or petroleum-based sample, the hydrocarbons are completely oxidized to CO2 and H2O and leave a clear aqueous solution with very small residual carbon content of <0.1%. This approach is exemplified in Figure 1a, which shows weighed samples of crude oil (mostly floating) loaded into high-purity quartz glass vials with doubly distilled strong acid–ultrapure peroxide mixture in a multisample numbered rack and readied for digestion. Figure 1b shows a completed high-pressure and temperature digestion just after the rack is raised from a previously sealed acid-resistant PTFE-lined stainless steel reaction chamber, which is prepressurized with inert gas before the digestion and then automatically ventilated upon completion and before extraction; the close-up image shows the undegassed digested sample (green solution containing oxides of nitrogen and carbon dioxide) in quartz vials with white laterally vented PTFE caps. Finally, Figure 1c shows subsequent degassed clear digest solutions in quartz glass PTFE-capped tubes, which are then placed in capsules ready for heating and dry-down procedures.



Figure 1: Single-reaction-chamber microwave digestion of crude oil, showing the various dissolution stages.
Digest solutions are then evaporated to near-dryness to remove strong acids and diluted with 2% HNO3 using a low dilution factor (8–10x). These solutions, which are basically free of carbon based spectral interferences are then introduced into the ICP-OES or ICP-MS system and analyzed using longer shelf life, commercially available aqueous calibration standards.
Compared to bomb mineralization or traditional closed-system rotor-based microwave systems, the advantage of the microwave-assisted strong acid wet digestion technique using the single reaction chamber include
- The multisample digestions are completed within 1 h compared to >24 h for bomb digestions.
- Multiple samples of different oils can be run at the same time under the same pressure–temperature conditions within a single reaction chamber.
- Pressure and temperature of the chamber wall and interior fluid chamber and microwave power is precisely controlled.
- High temperatures of 260 °C under controlled pressure can be achieved during glass vessel digestion for all samples.
- Sample sizes can be larger (for example, 0.6–1.2 g per vessel depending on rack sizes used), compared to 0.1 g for bombs or 0.25 g for typical rotor-based microwaves or closed-vessel bombs that are more limited to prevent venting and sample loss.
- Digestion vessel sizes can be varied (with multiple racks and vessel sizes possible within the single reaction cell) and can be used to influence ultimate measured sample sizes, dilution factors, and effectively will allow more control over all MDLs and MQLs.
This final point is particularly important when analyzing and quantifying very light oils, condensates, or naphtha, which collectively might have very low abundances of trace and ultratrace elements. The analysis of larger sample sizes is achieved by using larger vessels or combining sample digests from multiple vessels. They also ensure the likelihood of sample homogeneity.
Because the microwave energy is applied to all samples under the same temperature and pressure conditions, the system can sense highly exothermic reactions, pressure increases, and potential venting surges in real time in the single reaction vessel. It can then control power, which means it has the ability to abort potentially dangerous runs instantly if the outer chamber wall exceeds preset temperature limits or chamber pressure limits are exceeded. This leads to very safe digestions of combustible crude or fuel oils. The microwave system allows rapid digestion for increased throughput, allows minimal quantities of reagent use for clean chemistry, results in improved accuracy and precision because of minimal risk of contamination, minimizes sample loss of volatile elements, and leads to the destruction of organics because temperatures can exceed the boiling temperatures of reagents under pressure. Finally, the ability to use low-cost aqueous solution standards is important and cost effective, because the digestions can produce clear solutions for both crude and refined oils. This precise control over power, temperature, and pressure using this approach is seen in Figure 2, which shows a plot of the following metrics against time:
- programmed digestion temperature (red)
- maximum pressure (light blue)
- actual pressure (dark blue)
- maximum temperature of the reaction vessel outer wall (light green)
- actual temperature of the chamber outer wall (dark green)
- microwave power (black)


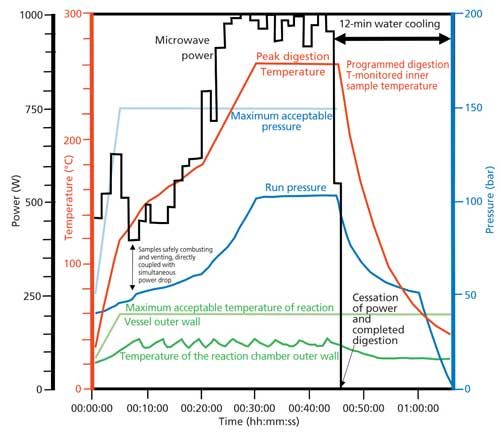
Figure 2: Plot of power, temperature, and pressure against time using a single-reaction-chamber microwave digestion system (refer to text for details).
ICP-OES and Triple-Quadrupole ICP-MS in Tandem for Routine Analysis
Crude oils and derivatives have elemental concentrations that range from 10–60 mg/g (1–6 wt %) to 1 pg/g, which means no single sample preparation methodology can be used with a single instrument that would have the dynamic range to analyze a comprehensive suite of elements detectable in crude oils without multiple preconcentration or added preparation steps, which would be very time consuming. However, by using the single prepared sample with two plasma spectrochemical techniques with different dynamic ranges (ICP-OES and ICP-MS), as used in our studies in tandem, the single sample preparation step outlined above can be used without the necessity of other time-consuming sample preparation steps. At a low 8.5x sample dilution used in our tests, ICP-OES is capable of analyzing higher abundance elements such as S, V, and Ni (and others) in some crude oils or refined derivatives and then aliquots of the same samples can be used to introduce into the ICP-MS system for analysis of low-abundance elements. It should be emphasized that low- and high-abundance elements are not consistent or predictable in crude oils, which can have a range of compositions even within the same subsurface petroleum-generating system depending on factors such as source rock depositional setting, thermal maturity, or biodegradation. For example, some heavy marine-sourced crude oils from euxinic basins may have >6% S by weight and 100–1000 µg/g V and Ni, which must be analyzed by ICP-OES, whereas others such as thermally mature shale oils, lucustrine oils, or condensates can have very low microgram-per-gram concentrations of S, V, and Ni that are better suited for the more sensitive triple-quadrupole ICP-MS.
The triple-quadrupole ICP-MS technology is a more recent development and includes an additional quadrupole before an octapole collision–reaction cell and the analyzer quadrupole. This first quadrupole acts as a simple mass filter to allow only the analyte masses to enter the cell, while rejecting all other masses. With all nonanalyte, plasma, and sample matrix ions excluded from the cell, sensitivity and interference removal efficiency is significantly improved compared to traditional collision–reaction cell (CRC) systems coupled with a single-quadrupole mass analyzer. In this very innovative CRC technology, different collision–reactive gases can be used to reduce the polyatomic interference through ion–molecule chemistry in the octopole cell, which is then used to pass all the product ions formed into the analyzer quadrupole and separate and select the mass or masses of interest. The benefit for analyzing and characterizing crude oil and petroleum samples is that notoriously difficult elements like silicon, phosphorus, and sulfur (and many others) can be determined at significantly lower levels and with higher accuracy than a conventional single-quadrupole instrument. This means that a larger elemental suite can be determined, which can potentially increase the knowledge used to identify the source and the origin of the crude oil sample.
In our comparative studies, we found that using a single-quadrupole ICP-MS and ICP-OES, we could routinely analyze and quantify up to ~47 elements with our digestion techniques, but with triple-quadrupole ICP-MS and ICP-OES we are able to routinely analyze and quantify up to 57 low- and high-abundance elements, mainly because of sensitivity increases and more control over the polyatomic spectral and other interferences. For example, with increasing focus on more stringent environmental management of pollutant emissions from heating oils and vehicle liquid fuels, sulfur, and other elements will be increasingly subject to environmental regulations and low-abundance elemental quantitation in fuel samples will be absolutely critical. This need will necessitate the use of the more advanced ICP-MS spectral reduction techniques to characterize the suite of elements at such low levels.
Recovery Tests and Accuracy
There are very few multielement certified natural crude oil standards, and typically they are certified for a very limited number of elements. We therefore used three approaches to examine recovery and accuracy: a certified synthetic organometallic multielement standard, a 57-element spike recovery test of a base oil blank, and a more limited test on reported NIST SRM oil values for the method developed.
Recovery tests of 20 elements were conducted on a commercially available Conostan (SCP Science) multielement organometallic standard digested with our microwave protocols and then analyzed by ICP-OES and ICP-MS. Figure 3 shows that the recoveries for all 20 elements are in the range of 95–107%.


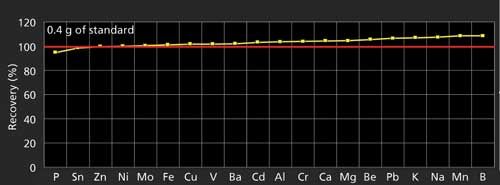
Figure 3: Recoveries of 20 elements in a multielement organometallic standard using microwave digestion and determined by ICP-OES or ICP-MS.
These results were augmented with our own tests in which 500 ng of 57 analyte elements were added to 400 mg of Conostan 75cSt base oil before microwave digestion to conduct a comprehensive recovery test for all elements routinely analyzed in the laboratory. These microwave-assisted digestion recovery results were superior to those obtained using all other digestion methods tested for the same suite of 57 elements and produced on average recoveries of 90-110%. Notably, there was good recovery of tested elements with known volatility issues, such as B, Be, Sn, As, Se, and Sb.
Accuracy Compared to a Standard Reference Material
Accuracy of any method is the agreement between a test result and certified reference values for comparison. Unfortunately, there is no natural oil SRM yet with extensive suites for elements certified to examine accuracy for the majority of elements analyzed. In this study, NIST SRM 1634c No.6 residual fuel oil was digested using our method and analyzed by the ICP-OES and ICP-MS techniques outlined for 57 elements. Results include five digestions run separately with two analyses for each digestion. The results shown in Figure 4 are a log-scale spidergram of concentrations (in nanograms per gram) for five digestions at different times with two analyses of each digestion, making 10 replicates in total.


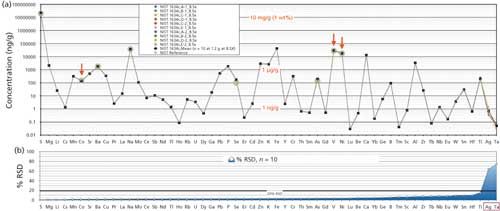
Figure 4: Log-scale spidergram of concentrations (ng/g) of 10 replicated analyses of 57 elements measured in NIST SRM 1634C fuel oil, determined by ICP-OES or ICP-MS, together with the RSD of each measurement.
The precision data underneath the spidergram shows the precision (% RSD) for all elements. It can be seen that all elements were well below 20% RSD even though some elemental abundances were near their MQL. Exceptions include Ag and Ta (far right), both with concentrations less than 1 ng/g, which showed far greater scatter with high RSD. Elemental concentrations ranged widely from more than 2% (20 mg/g) by weight for S and 1–40 µg/g for V, Ni, K, and Na requiring quantitation by ICP-OES, to sub nanogram-per-gram concentrations requiring the remainder of elements to be quantified by triple-quadrupole ICP-MS.
The highlighted green circles represent superimposed NIST “certified” values for Co, Ni, and V as well as “reference” values for As and Se, and “information” values for Ba, Na, and S. The accuracy assessment of our mean achieved values could only realistically be compared to NIST certified values shown in Table I, which include Co (9.4%), Ni (1.4%), and V (2.4%). Noncertified values are also listed in Table I for further information. Compared to NIST noncertified “reference” values reported for As and Se, elements known to have volatility issues were somewhat higher in our analyses (27% and 42%, respectively) than NIST values although precisions were <4%. Testing by Wondimu and Goessler (8) based on a closed-system rotor-based microwave digestion on the same standard also reported similarly higher values than “reference” values listed by NIST with precisions <5%. The reason is unclear, but higher values could indicate more complete recovery and higher measurement values are associated with closed microwave digestions, or may be related to the differences in analytical techniques used. All “information” values of Ba, Na, and S compare well with the NIST results reported (<10%). Note that many of the NIST analytical methods in Table I differ from the two ICP instrumental techniques and digestion procedures used in our evaluation. Developments and further definition of “true” values for noncertified “reference” and “information” values will require further inter-laboratory comparisons, and NIST statistical testing to allow the potential of future compliance with NIST certification criteria.
It should also be emphasized that the NIST 1634c SRM was chosen for this study because of the comparatively larger number of elements reported by NIST (eight in total), which are measurable by our chosen ICP techniques. The achieved mean concentration results, RSDs, and accuracy estimates compared with NIST reported certified, reference, and informational values are shown in Table I.
Within-Laboratory Precision, Repeatability, and Reproducibility
Within-laboratory precisions of our analyses can be estimated on the basis of the 10 replicate determinations of 57 elements for NIST SRM 1634c. As previously mentioned, the concentrations and the % RSD or variance for each element are plotted in Figure 4. The % RSD of the mean of each element is shown (10 analyses of the SRM) with elements listed in order of increasing RSD value. Analyses of this standard involved five separate microwave digestions (A–E) over time with replicate analysis of each digestion (for example, A1 and A2). Results are plotted as concentrations in nanograms per gram. Of the 57 elements measured, 42 elements had RSDs ranging from 0.7% to 4.9%, 12 elements had RSDs ranging from 5.2% and 10.0%, and two elements had RSDs ranging from 10.4% to 15.3%, meeting our imposed RSD limits of <20% of acceptability even at sub-nanogram-per-gram levels. Two elements, Ag and Ta, were <1 ng/g in abundance and showed more significant heterogeneity, with RSDs of 63.7% and 74.5%, respectively, and did not meet stricter quantitation criteria. Whereas RSDs are generally low for the 55 elements quantified, the exceptions on these runs were Ag and Ta. Poorer Ag precision sometimes observed for repeat measurements of samples and for NIST 1634c may be a consequence of the low concentration, which is at or near the MQL of 0.03 ng/g and the fact that Ag is also known for its tendency to form matrix-dependent salts over time in solutions, especially at low concentrations. Both factors could contribute to the variability observed. The higher RSD for Ta may also result from matrix-dependent solubility issues, because the element has been observed to drop out of solution over time (~4 weeks) in prepared low-abundance multielement standard solutions. Investigations to better optimize these elements and procedures are underway, although variance is commonly lower in other samples tested.
Poor repeatability is a well-known problem for a number of analytical techniques used to analyze digests of crude and fuel oils (1). However, in our duplicate digestions and replicate analyses presented here, 55 of the elements that are significantly above the MQL show good within-laboratory RSDs of 0.7-15%. We have also observed the same consistency in other NIST crude oil standards with many replicate digestions and with the natural crude oils analyzed for up to 57 element when analyses were repeated by various laboratory investigators, at various times, and with different reagent batches. In addition to within-laboratory repeatability tests, future studies will involve interlaboratory comparisons to examine the ruggedness of methods. Interlaboratory precision predictions can, however, be made from the Horwitz empirical method and reproducibility curve (11). Based on our repeatability tests and intermediate method precision data, it can be predicted that the overall method should be robust in interlaboratory comparisons for most all of the analytes measured.
Examples of Crude Oil Fingerprints
In Figure 5, we show two examples of natural crude oil fingerprinted by our analytical method in comparison with NIST 1634c fuel oil results on a log-scale spidergram. These three analyses illustrate distinct fingerprints for each oil with many elements varying by more than 2–3 orders of magnitude. One of the natural oils was relatively lighter and generated in a subsurface lacustrine source rock environment in Sumatra, while the other was a heavier oil generated in a marine euxinic environment off the coast of California.


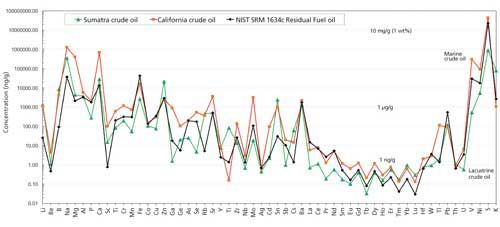
Figure 5: Results obtained using the developed method for fingerprinting two natural crude oils-one from a lacustrine source rock in Sumatra (green) and the other from a marine euxinic environment off the coast of California (orange)-compared with the NIST SRM 1634c residual fuel oil (black).
All element abundances are at or above MQL. They are readily distinguished by distinct ratios and abundances of V and Ni and other elements that can allow oil–oil or oil–source rock correlations to be made by oil exploration scientists. We are also able to distinguish crude oils from different wells in the same fields by this fingerprinting technique. In all, there are 57 element abundances and more than 3000 ratios that can be utilized by upstream explorationists for fingerprinting, oil–oil and oil–source rock correlation, organic matter typing, depositional environment typing, and maturation and biodegradation studies of oils or source rock organic matter (12–16). As in crude oil organic geochemistry, elemental abundances and ratios can also be useful in reservoir management, production allocation, and transportation and flow assurance engineering (17,18). In addition, downstream refiners have needs to characterize natural or comingled crude oils, feedstocks, and refined products to identify catalyst poisons and corrosive metal species, to control refinery costs, maintenance schedules, and for quality assurance purposes to meet more stringent regulatory requirements and industry standards for refined products (19).
Summary
Recent significant developments in microwave digestion technologies and ICP-MS analytical instruments show significant promise in resolving many of the past issues related to the rapid multielement analysis of crude oil and derivative products. Whereas dilute-and-shoot industry-standard methods will continue to have significant applications, we regard the oil chemical fingerprinting analytical methods developed here as a potential pathway forward to more-routine, lower-cost multielement analysis with a more comprehensive elemental list if the industry is to meet progressively more complex and challenging needs. These more-extensive target analytes include those currently well-accepted and designated as important and useful to the upstream and downstream petroleum industries, as well as low-abundance, poorly documented elements (such as rare earths) whose routine testing is yet to be fully realized. This more-comprehensive elemental list may lead to more sophisticated applications of elemental analysis for crude oil and derivatives, with benefits extending from exploration and production to refining, product monitoring, and regulatory and compliance efforts. With the optimization in sample preparation and the types and numbers of dedicated instruments required for more complete high- to trace-level elemental characterization, the lower expenditure will translate into an overall reduction in operational and discovery costs.
Disclaimer
Robert Thomas, the editor of the “Atomic Perspectives” column would like to emphasize, that although he contributed to the writing of this month’s column, he does not endorse any of the commercial products mentioned herein.
References
- C. Duyck, N. Miekeley, C.L.P. Silveira, R.Q. Aucélio, R.C. Campos, P. Grinberg, and G.P. Brandão, Spectrochim. Acta, Part B62, 939–951 (2007).
- J.F. Casey and Y. Gao, “Triple Quadrupole ICP-MS and ICP-OES Fingerprinting of Up to 57 Trace and Minor Elements in Crude Oil Using a Single Sample Preparation Method,” available at: http://cen.acs.org/media/webinar/agilent_102015.html (2015).
- J.F. Casey, Y. Gao, and W. Yang, “Analysis of Low Abundance Trace Metals and 50V/51V Isotopes in Crude Oils: New Methods for Characterization and Exploration” presented at the Goldschmidt 2015 conference, Prague, 2015.
- C. Duyck, N. Miekeley, C.L.P. Silveira, and P. Szatmari, Spectrochim. Acta, Part B57, 1979–1990 (2002).
- C.J. Lord III, Anal. Chem. 63(15), 1594–1599 (1991).
- G.S. Ortega, C. Pécheyran, G. Hudin, E. Marosits, and O.F. Donard, Microchem. J. 106, 250–254 (2013).
- T.A. Yasnygina, S.V. Rasskazov, M.E. Markova, A.E. Zharov, Y.M. Malykh, E.V. Saranina, and N.N. Fefelov, Russian Journal of Pacific Geology9(2), 109–119 (2015).
- T. Wondimu and W. Goessler, Bull. Chem. Soc. Ethiop.14.2, 99–113 (2000).
- C.N. Duyck, N. Miekeley, T.C.O. Fonseca, P. Szatmari, and E.V. dos Santos Netob, J. Braz. Chem. Soc.19(5), 978–986 (2008).
- J.S.F. Pereira, D.P. Moraes, F.G. Antes, L.O. Diehl, M.F.P. Santos, R.C.L. Guimarães, T.C.O. Fonseca, V.L. Dressler, and É.M.M. Flores, Microchem. J.96(1), 4–11 (2010).
- W. Horwitz, L.R. Kamps, and K.W. Boyer, J. Assoc. Off. Chem.63, 1344 (1980).
- M.D. Lewan, Geochim. Cosmochim. Acta48(11), 2231–2238 (1984).
- A.J.G. Barwise, Energy Fuels4(6), 647–652 (1990).
- R.H. Filby, Geological Society, London, Special Publications 78, 203-219 (1994).
- F. Galarraga, K. Reategui, A. Martinez, M. Martinez, J. Llamas, and G. Márquez, J. Pet. Sci. Eng.61, 9–14 (2008).
- R. Nakada, Y. Takahashi, G. Zheng, Y. Yamamoto, and H. Shimizu, Geochem. J.44(5), 411–418 (2010).
- M.A. McCaffrey, H.A. Legarre, and S.J. Johnson, AAPG Bulletin80, 898–913 (1996).
- R.J. Hwang, D.K. Baskin, and S.C. Teerman, Org. Geochem. 31, 1463–1474 (2000).
- J.G. Gary and G.E. Handwerk, Chemical Processing and Engineering, 4th. Ed. (Marcel Dekker Inc., New York, 2001).
- W. Yang, J. F. Casey, and Y. Gao, Fuels (in review).
- J.F. Casey. Y. Gao, W. Yang, and K.A. Bissada, Geological Society of London, special edition “From source to seep: Geochemical applications in hydrocarbon systems,” in review.



Robert Thomas is principal of Scientific Solutions, a consulting company that serves the application and writing needs of the trace element user community. He has worked in the field of atomic and mass spectroscopy for more than 40 years and has written over 80 technical publications including a 15-part tutorial series on ICP-MS. He recently completed his third textbook entitled Practical Guide to ICP-MS: A Tutorial for Beginners. He has an advanced degree in analytical chemistry from the University of Wales, UK, and is also a Fellow of the Royal Society of Chemistry (FRSC) and a Chartered Chemist (CChem).



John F. Casey is a Professor in the Geology Department at the University of Houston in the Earth and Atmospheric Sciences Department. He serves as the Director of a University core facility, the Center for Advanced Analytical Geochemistry (CAAG). He has worked in the field of trace element and isotopic geochemistry of terrestrial and marine geological materials since the early 1980s. He is the author of more than 100 peer-reviewed articles in the geosciences.



Yongjun Gao is a Research Associate Professor at the University of Houston in the Department of Earth and Atmospheric Sciences. He has 15 years of experience in conducting geochemistry research in various fields and has published more than 40 peer reviewed research articles.



Weihang Yang is a PhD candidate and teaching assistant of Geology in the Earth and Atmospheric Sciences Department at the University of Houston.




Newsletter
Get essential updates on the latest spectroscopy technologies, regulatory standards, and best practices—subscribe today to Spectroscopy.






Mediterranean Origins of Red Coral Artifacts in Xinjiang Reveal Ancient Silk Road Trade Links
June 26th 2025A new study published in Minerals reveals that red coral artifacts unearthed in Xinjiang’s Shengjindian cemetery originated from the western Mediterranean, highlighting early Silk Road trade and long-distance cultural exchange during the Han Dynasty.

The Role of ICP-OES in Analyzing the Metal Content in Pet Food
June 19th 2025Because the United Arab Emirates is seeing an increase in pet ownership, the quality of both dry and wet pet food is undergoing greater scrutiny to ensure its safety and efficacy. Lucy Semerjian, who works as a Chair and Associate Professor in the Department of Environmental Health Science at the University of Sharjah in Sharjah, United Arab Emirates, recently explored this topic in a recent paper
Pet Food in the United Arab Emirates: An Interview with Lucy Semerjian
June 18th 2025A recent study conducted in the Journal of Food Composition and Analysis examined the concentrations of ten metals in 52 commercially available wet and dry cat food samples, assessing their compliance with U.S. and European pet food safety standards. The lead author of this study, Lucy Semerjian, recently sat down with Spectroscopy to discuss the findings of her study.


New Study Finds Elevated Metal Levels in Some Cat Foods Sold in Sharjah
May 27th 2025A new study published in the Journal of Food Composition and Analysis by researchers at the University of Sharjah reveals that while most cat foods sold in Sharjah meet international safety standards, some contain elevated metal levels, prompting calls for stricter regulation and quality control to protect pet health.

