Raman Spectroscopy and Polymorphism
The differentiation of polymorphs is important, particularly in the pharmaceutical industry. We demonstrate the practicality of using Raman spectroscopy to differentiate crystal forms for polymorph characterization and screening, and explain aspects of chemical bonding and solid state structure that affect the Raman spectra of crystal lattice vibrational modes.
Single crystals can be formed that have different unit cells. Crystal polymorphism is the formation of a compound in various crystallographic forms. The differentiation of polymorphs is important, particularly in the pharmaceutical industry. Raman spectroscopy has emerged as a complementary method to X-ray diffraction, the "gold standard" for characterization of crystal structure. Different crystal forms of ionic or covalent solids and molecular crystals can be differentiated through Raman spectroscopy. The Raman spectra of molecular crystals consist of bands attributable to external and internal crystal lattice vibrational modes. We discuss these aspects of chemical bonding and solid state structure that affect the Raman spectra of crystal lattice vibrational modes, and demonstrate the practicality of using Raman spectroscopy to differentiate crystal forms for polymorph characterization and screening.
Polymorphism is a term used in various scientific disciplines with different meanings. Here, we use it to describe the multiplicity of phases or crystal structures of a single compound. Crystal polymorphism is the formation of a compound in various crystallographic forms; that is, single crystals can be formed that have different unit cells. The differentiation of polymorphs is important, particularly in the pharmaceutical industry. The crystal form of an active pharmaceutical ingredient can affect its chemical or physical stability, and its dissolution rate. Consequently, the crystal structure of a pharmaceutical compound can have a profound effect on its efficacy and potency. Of course, to analyze crystal structure, X-ray diffraction is considered the "gold standard" for characterizing and differentiating crystal forms of compounds. However, once different phases of a chemical compound have been confirmed or newly identified by X-ray diffraction, those same samples can be used to generate reference Raman spectra of that compound's specific crystal forms. Raman spectroscopy is often found to be experimentally more convenient to use than X-ray diffraction for polymorph characterization or screening. Reference Raman spectra of a compound's polymorphs can be reliably used to confirm crystal forms, or even to identify new ones in a research setting or in polymorph screening. The most important aspect of Raman spectroscopy when applying it to polymorph analysis is spectral resolution. That is consistent with the fact that high spectral resolution is also required to differentiate crystal forms by X-ray diffraction. The Raman spectrometer must have spectral resolution sufficient to resolve the smallest differences in energies of the crystal lattice vibrational modes, which are affected by changes in molecular interactions arising from different unit cell structures or configurations of the molecules, or formula units within the unit cell. Just as small differences in a compound's crystal structure lead to small differences in the X-ray diffraction pattern, the Raman spectra of crystals with small differences in bond lengths or crystal spacing will manifest small differences in the Raman peak positions. Hence, just as high resolution X-ray diffraction is needed to resolve polymorphs, so too is high spectral resolution required to differentiate crystal forms by Raman spectroscopy.
Many spectroscopists are chemists, and therefore most likely learned vibrational spectroscopy as molecular spectroscopy. If you are a chemist, you learned about the normal vibrational modes of discrete molecules, as opposed to solid state materials, and their Raman or infrared activity based upon molecular spectroscopic selection rules. Raman spectra of compounds in the liquid or vapor phase consist of narrow bands whose widths depend upon the degree of chemical interaction between the molecules. The weaker the chemical interaction, the narrower the band will be. In fact, the bands of a compound in the vapor phase will be narrower than those of the same compound in the liquid phase for the very reason that the molecular interactions are weaker (1). The broad infrared absorption and Raman bands of water demonstrate the effect of hydrogen bonding and the result of different degrees of chemical interaction between the molecules in a neat liquid. The breadth of the O-H stretching modes in particular is a manifestation of the distribution of vibrational energy states as a result of these many and different chemical interactions. Those same molecular interactions can affect the vibrational spectrum of a compound in the solid state, and are dependent upon how the molecules are configured or oriented in the unit cell.
Interpreting the Raman spectra of compounds or materials in the solid state requires the knowledge of concepts and mathematical treatments other than those of molecular spectroscopy. The purpose of this installment of "Molecular Spectroscopy Workbench" is to demonstrate the use of Raman spectroscopy for differentiating polymorphs, and to explain the underlying bases for that capability. In particular, we examine and discuss the low and high frequency regions of a Raman spectrum, explaining the different types of crystal lattice vibrational modes from which they originate and comparing their suitability for distinguishing crystal forms. To appreciate why Raman spectroscopy is sensitive to crystal structure, we need to understand the origins of Raman scattering from solid state materials. Raman scattered photons are generated in solid state materials through the creation or annihilation of phonons which correspond to Stokes and anti-Stokes Raman scattering, respectively. A phonon is defined as a crystal lattice vibrational wave propagating through the crystal arising from repetitive and systematic atomic displacements. It should also be understood that these displacements are quantized vibrations of the atoms in the crystal lattice and are travelling waves. The phonon has the characteristics of a travelling wave insofar as it has a propagation velocity, wavelength, wave vector, and frequency. It is important to note that all of the peaks in a Raman spectrum of a crystalline solid are attributed to phonons, and not only those at low energy or Raman shift. P.M.A. Sherwood makes that point clear in his fine book Vibrational Spectroscopy of Solids: "All crystal vibrations involve the entire lattice and are thus lattice vibrations (a term sometimes unfortunately only applied to external vibrations) and such vibrations can be considered as a wave propagating through the crystal lattice" (2). Unlike the normal vibrational mode of an individual molecule, a phonon is a crystal lattice vibrational wave travelling through the crystal. This is why a mathematical treatment of the vibrational motions of the crystal's atoms must take into account the medium through which the phonon is propagating, and why the energy of the phonon depends upon the configuration of the atoms and chemical bonds within the unit cell.
In chemistry courses, we learn about Raman scattering through the interaction of light with molecules. A molecule, initially in the ground state, interacts with an incident photon driving the molecule to a virtual energy state whereupon it drops to the first excited vibrational state and emits a photon (Stokes Raman scattering). The energy difference between the incident and scattered photons is equal to the energy difference between the ground and first excited vibrational states. Had the molecule initially been in the first excited vibrational state and ended in the ground state, a quantum of energy would have been transferred from the molecule to the scattered photon, and its energy would have been greater than that of the incident photon (anti-Stokes Raman scattering). Hence, one can observe the Raman spectrum at either longer (Stokes) or shorter (anti-Stokes) wavelengths relative to that of the incident monochromatic laser beam.
When dealing with gases or liquids, it is appropriate to speak of the interaction of a photon with individual molecules. However, very often we deal with solid state materials for which there may be no molecular species, such as titanium dioxide (TiO2), silicon (Si), carbon (C, graphene or diamond), or calcium carbonate (CaCO3). Raman spectroscopy of solid state materials involves the inelastic scattering of light by phonons, quanta that have the energy of crystal lattice vibrations. The Stokes and anti-Stokes Raman scattering consists of the generation or annihilation of a phonon in the solid, respectively. In crystalline materials, the phonons can be understood as crystal lattice vibrational modes whether the crystal is a covalent or ionic solid or a molecular crystal such as TiO2, barium fluoride (BaF2), or water (H2O, ice), respectively.
When speaking of Raman spectra of solid state materials, some spectroscopists will describe certain bands as phonons, and others as molecular vibrations. Strictly speaking, this is not correct. All of the bands in a Raman spectrum of a solid arise from phonons. A more appropriate distinction is to speak of external and internal crystal lattice vibrational modes when speaking of solid state Raman band assignments, particularly for molecular crystals (2). An external crystal lattice vibrational mode can be thought of as the collective motion of molecules as a whole, such as whole water molecules moving collectively in an ice crystal. Some low energy Raman bands are a result of shear or interlayer breathing modes of the crystal layers. The shear modes can be pictured as atomic or molecular layers moving antiparallel to each other within their respective planes, whereas the breathing modes involve the layers moving away from and towards each other. The external crystal lattice vibrations are generally of low energy, and, of course, would be absent from the liquid or gas spectrum of the material because of the absence of long range translational symmetry that is present in a crystal. The internal crystal lattice vibrational modes arise from the coupling through the crystal of the local vibrational modes observed for the molecular species. We can think of these types of phonons as collective local modes modified through coupling with other molecules in the crystal and affected by their arrangement in the crystal lattice. Their Raman shifts are often similar but not identical to those of the molecular (liquid or gas) spectrum.
As you may expect, the external lattice vibrational modes or low energy phonons are very sensitive to crystal structure. That is why the low frequency region of the Raman spectrum has proven so useful in the pharmaceutical industry for the characterization and screening of polymorphs. In addition, Raman spectra of low energy phonons are dependent upon the crystal orientation with respect to the incident laser polarization as are the higher energy internal crystal lattice modes. Perhaps less well known is the sensitivity of the external modes to molecular interactions, such as hydrogen bonding or even the weaker van der Waals forces between molecules, or formula unit layers within the crystallographic unit cell. This sensitivity to the chemical interactions results in the energies of the external modes, and therefore their Raman peak positions, being strongly affected by the configurations of molecules or formula units within the unit cell. We present and discuss the Raman spectra of TiO2, paracetamol, and carbamazepine to demonstrate the merits of using Raman spectroscopy to differentiate crystal forms for polymorph characterization and screening.
Anatase and Rutile TiO2
There are multiple crystalline phases of TiO2, and the ones with which you may be the most familiar are anatase (tetragonal), rutile (tetragonal), and brookite (orthorhombic). Of these three, the two most commonly encountered in an industrial setting are anatase and rutile. Anatase belongs to the crystal class of D4h, with four formula units per crystallographic unit cell. The correlation method for vibrational selection rules predicts six Raman active modes (A1g + 2 B1g + 3 Eg) for anatase TiO2 (3,4). Rutile TiO2 also belongs to the crystal class of D4h, but has two formula units per crystallographic unit cell. The correlation method predicts four Raman active modes (A1g + B1g + B2g + Eg) for rutile TiO2 (3,5). Clearly, then, one should in principle be able to differentiate the anatase and rutile phases of TiO2 by Raman spectroscopy just by applying the Raman polarization selection rules to identify the symmetry species of Raman bands and by the total number of bands observed in the Raman spectrum. Nevertheless, the application of group theory and Raman polarization selection rules for the purpose of identifying or differentiating crystal forms is not always straightforward.
Group theory and the correlation method are beneficial for identifying Raman active modes and assigning a symmetry species to a particular band, but they do not predict the Raman scattering strength of the individual crystal lattice vibrational modes. Furthermore, second order modes can also appear in the Raman spectrum, thereby complicating the assignment of symmetry species and the total count of bands attributed to fundamental lattice vibrational modes appearing in a spectrum. The Raman spectra of anatase and rutile TiO2 are shown in Figure 1 (These spectra and all the others shown in this publication were acquired using 532 nm excitation and a long working distance 50X Olympus microscope objective). The rutile spectrum consists of four bands as predicted by the correlation method. The bands at 143, 445, and 610 cm-1 have been assigned to the B1g, Eg and A1g symmetry species, respectively (6). However, the broad band at 241 cm-1 has been attributed to second- order or two-phonon Raman scattering, and so cannot be counted among the four fundamental modes predicted by group theory (6). Where then is the fourth fundamental mode? Although barely discernable in the rutile TiO2 spectrum, there is a very weak band at 828 cm-1 which has been assigned to the B2g symmetry species. All four of the fundamental lattice vibrational modes are now accounted for. You will also notice a broad shoulder at approximately 707 cm-1 which can be attributed to two-phonon Raman scattering. Although one might have expected a simple spectrum consisting of four bands based upon group theory analysis and the correlation method, a total of six bands are present, two of which are due to second order Raman scattering. The lesson to be learned is that the interpretation of even as simple a spectrum as that of rutile TiO2 is not straightforward, and we are aided in our assignments and interpretation by previously published work. It is helpful to apply group theory and the correlation method when performing crystallographic studies by Raman spectroscopy, but be sure to consult previously reported results in order to perform a thorough study and make the most accurate band assignments and interpretations of spectra.


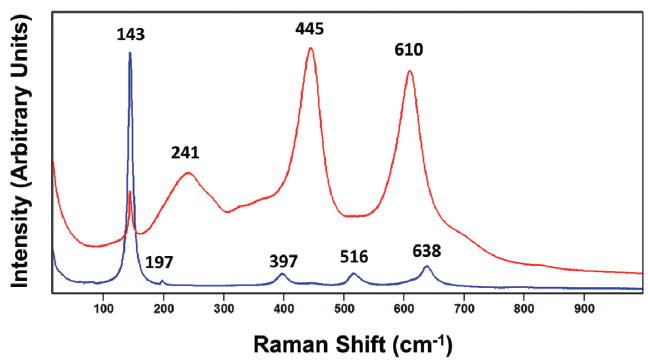
Figure 1: Raman spectra of the rutile (red) and anatase (blue) crystalline forms of titanium dioxide (TiO2).
The total number of bands observed in the anatase TiO2 spectrum of Figure 1 is only five, whereas six are predicted. The bands and their symmetry species assignments are 143 cm-1 (Eg), 197 cm-1 (Eg), 397 cm-1 (B1g), 516 cm-1 (B1g), and 638 cm-1 (Eg) (6). Those five assignments account for all but one A1g band. According to Tompsett and coworkers, the missing A1g band appears at 507 cm-1 and can only be resolved from the 516 cm-1 (B1g) band at temperatures below 73 K (6). And so, with a little work involving group theory, the correlation method and study of the literature we are able to assign the Raman bands, and differentiate the anatase and rutile forms of TiO2 by their distinctly different spectra. Taken together, we see that the key to differentiating these two polymorphs is the peak positions and relative intensities of the two crystal forms. However, as we will see when discussing the Raman spectra of the molecular crystals paracetamol and carbamazepine, one must be cautious and not over interpret the significance of different relative intensities. The analysis of different grains of the same crystal form of a compound can produce spectra of vastly different relative intensities, which can lead the spectroscopist to incorrectly conclude that the grains are of different crystal forms. The differences in peak positions are far more reliable than different relative intensities for differentiating crystal forms.
It is also important to keep in mind that samples of a single compound may not necessarily consist of a single phase or crystal form. One may find, even on a scale of only a few micrometers, mixtures of crystalline phases. For example, a spectrum shown in Figure 2 was acquired from a grain that is nominally anatase TiO2 but its spectrum is comprised of bands attributable to both the anatase and rutile phases. In spite of the fact that the beam diameter is less than several micrometers, the spectrum consists of contributions from two phases. Clearly, crystallographic domains or grain sizes can be very small, and so one must be aware of this fact even when performing micro-Raman spectroscopy. The bands at 143, 197, 397, 516 and 638 cm-1 are those expected for anatase TiO2. However, the bands at 445 and 610 cm-1 very clearly correspond to those of our reference rutile phase, thereby leading us to conclude that the sample is not purely anatase, in spite of the fact that it was labeled as only anatase. In fact, with our magnified intensity scale in Figure 2, one can detect weak contributions at 445 and 610 cm-1 in our anatase TiO2 reference spectrum, thereby indicating that our anatase reference material may not be entirely pure. Another possible interpretation of the middle spectrum in Figure 2 is that it may indicate the presence of the brookite phase of TiO2. However, the brookite phase TiO2 spectrum has many more bands than the middle spectrum, and the peak positions are not a good match (6). Therefore, we can reasonably conclude that the sample is primarily anatase TiO2 with some rutile TiO2 present.


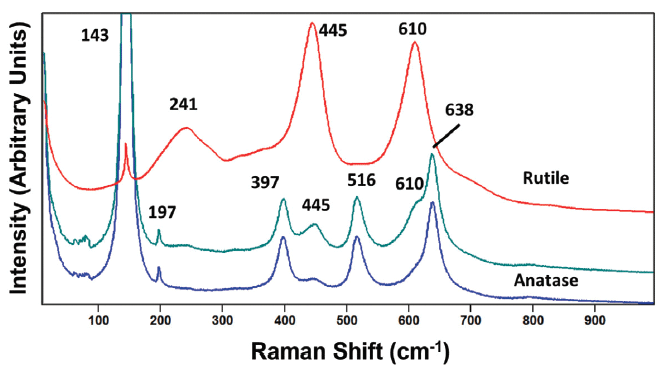
Figure 2: Raman spectrum obtained from a grain of anatase titanium dioxide (TiO2) with an impurity phase of rutile present (green). The Raman spectra of reference rutile and anatase phases are shown in red and blue, respectively.
To further emphasize the need to be aware of the presence of multiple forms or impurity phases, consider the spectrum in Figure 3 from a sample labeled as rutile TiO2. As expected, the rutile bands at 241, 445, and 610 cm-1 are present. However, we see unexpectedly strong Raman scattering at 143 cm-1, and bands at 197, 397, 516, and 638 cm-1, which can be attributed to the anatase form of TiO2. Of course, the contributions of the signal strengths of the two phases are directly proportional to their relative amounts, and it would appear that the spectral contribution of the impurity phase is greater here than that in Figure 2. However, caution is necessary if one is to use the relative signal strengths to perform even a semi-quantitative analysis of a sample consisting of multiple phases. Reference spectra of the pure phases would be required, and the optical experimental conditions for sample laser illumination and light collection should be comparable, if not identical, in order to accurately perform a semi-quantitative mixture analysis. Also, don't forget the importance of the sample shape and size. A single crystal with a flat surface oriented in a particular plane relative to the incident laser polarization will yield a spectrum of quite different relative intensities compared to that of a polycrystalline grain of the same crystalline phase. It would be very difficult to perform an accurate quantitative mixture analysis if the grain morphology closely approximated that of a single crystal.


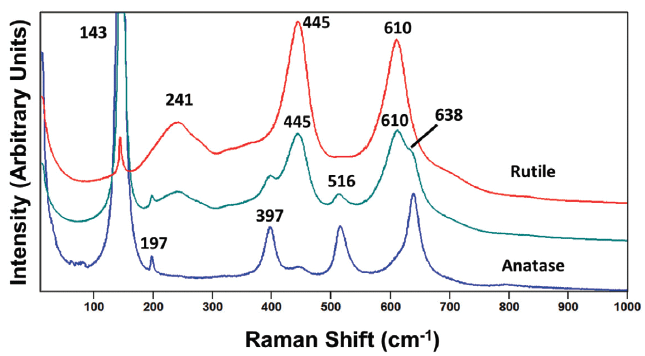
Figure 3: Raman spectrum obtained from a grain of rutile titanium dioxide (TiO2) with an impurity phase of anatase present (green). The Raman spectra of reference rutile and anatase phases are shown in red and blue, respectively.
Paracetamol Forms I and II
Whereas TiO2 is a covalent solid and no individual TiO2 molecules exist at standard temperature and pressure, the common pain reliever paracetamol is a molecular crystal consisting of molecules in the crystallographic unit cell. Paracetamol is a solid at room temperature, but, if heated and melted, will form a liquid consisting of paracetamol molecules. That is not the case for TiO2. There are significant differences between the vibrational spectra of ionic or covalent solids and those of molecular crystals. The Raman spectrum of a molecular crystal will be similar, but not identical, to that of the molecular (liquid or vapor phase) spectrum of the same compound. Those bands in the Raman spectrum of a molecular crystal that are similar to those found in the molecular spectrum of the same compound in either the liquid or vapor phase are attributed to internal phonon modes. The designation internal phonon mode arises from the approximation of those crystal lattice vibrational modes being associated with the functional groups or structural units (tetrahedra or octahedra) of the molecule and their stretching or angle bending of local bonding in the molecule. When a crystal is formed, the atom–bond or bond–bond molecular interactions within the unit cell perturb the energetics of the bonds, and thereby affect the vibrational frequencies of those internal phonon modes. The energies of even the internal phonon modes can be affected by near neighbor chemical interactions depending upon the arrangements of the molecules within the unit cell. Consequently, crystal structures with different configurations of the molecules within the unit cell will manifest different molecular interactions, thereby affecting the energies of the crystal lattice vibrational modes, including the internal phonon modes. That is the reason for the differences in Raman spectra of polymorphs of a molecular crystal. The energies of the crystal lattice vibrational modes depend upon the configuration of the molecules within the crystallographic unit cell, and, in particular, their molecular interactions.
As discussed in the introduction, the external crystal lattice vibrational modes are those involving the collective oscillation of entire molecular species within the unit cell and propagating through the crystal. These collective oscillations of entire molecules can consist of shear modes, so-called interlayer breathing modes (wherein the molecules move towards and away from each other), and bending modes (where the angle between molecules varies). As you might expect, these lattice vibrational modes are at low frequency, typically less than 100 cm-1, and as low as 5 cm-1, because of the higher mass of the collective movements of entire molecules within the unit cell. This region of the vibrational spectrum corresponds to the terahertz frequencies when performing infrared absorption spectroscopy. The frequencies of the external vibrational modes are very sensitive to the molecular arrangements within the unit cell. Consequently, the external crystal lattice vibrational modes are generally the most sensitive to crystal form, and the most beneficial for differentiating polymorphs. We will compare the efficacy of the internal and external crystal lattice vibrational modes for characterizing and differentiating polymorphs in the following discussions of Raman spectra of paracetamol and carbamazepine.
Spectra consisting of bands due to external and internal crystal lattice vibrational modes of the fingerprint region acquired from two forms of paracetamol are shown in Figure 4. Note how similar the spectra of forms I and II are at Raman shifts greater than 200 cm-1, whereas they appear quite different between 10 and 200 cm-1. Of course, what we have in mind in making that statement is the apparent similarity of the approximate band positions and the relative intensities of the two spectra. One might therefore incorrectly conclude from Figure 4 that merely examining the low frequency portion of the spectrum is all that is necessary to distinguish polymorphs, and that the high frequency region is not helpful in differentiating crystal forms. However, viewing the spectra of forms I and II in this fashion for purposes of comparison can be a little deceptive. We shall see that, when viewed on a different scale, the actual peak positions of forms I and II are not exactly the same for the two forms, and that the bands from internal phonon modes can be effectively used to differentiate polymorphs.


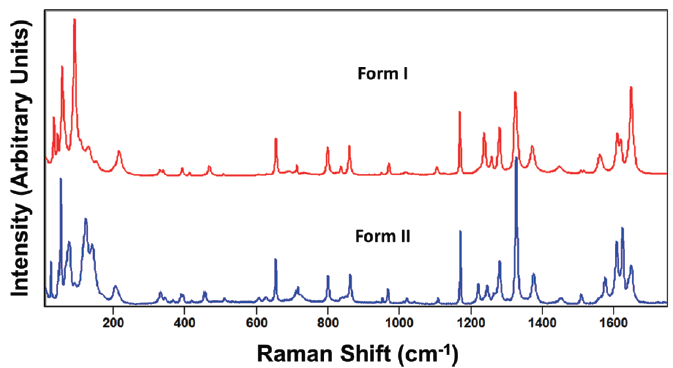
Figure 4: Raman spectra obtained from grains of paracetamol form I (red) and form II (blue).
An important consideration in spectral interpretation is the optical sampling of the spectral acquisition. This is critically important particularly if performing micro-Raman spectroscopy where the orientation of individual sample grains relative to the direction and polarization of the incident laser beam can have significant effects on the acquired spectrum. For example, compare the spectra shown in Figure 5 acquired in the same spectral region of 10 to 1750 cm-1 from different grains of paracetamol form I. These spectra, although acquired from different grains of the same crystal form, appear more dissimilar than the spectra obtained from the two different crystal forms shown in Figure 4. If one were not careful, one might incorrectly conclude that the spectra in Figure 5 were acquired from two different crystal forms. Likewise, the spectra shown in Figure 6 were acquired from different grains of form II paracetamol. The spectra appear quite similar at Raman shifts greater than 200 cm-1 as we would expect. However, below 200 cm-1 the band structures appear quite different, and one might incorrectly conclude that the spectra were acquired from different crystal forms.


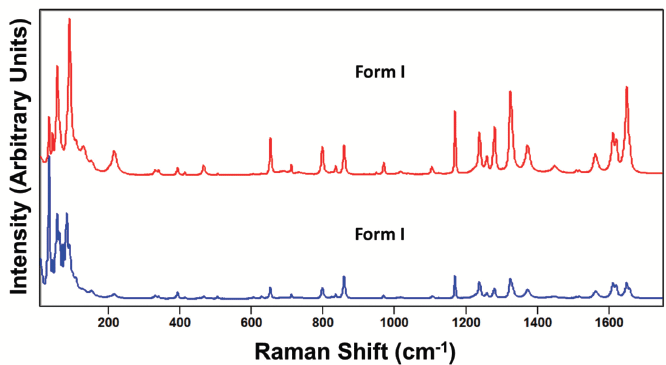
Figure 5: Raman spectra obtained from different grains of paracetamol form I.
So where does this leave us? How are we to confidently apply Raman spectroscopy to characterize and differentiate polymorphs of molecular crystals if the spectra obtained from different grains of the same crystal form can appear different? This is where the spectral resolution of the Raman spectrometer is so important. We start by analyzing and comparing the peak positions of the Raman spectra of paracetamol forms I and II at the higher frequencies of the fingerprint region shown in Figure 7. The spectra are indeed similar as one would expect, because they are spectra from molecular crystals of the same compound. However, they do not have exactly the same peak positions because the energies of their corresponding phonons are different due to slightly different crystal structures. For example, there is a 6 cm-1 difference between the 1619 and 1625 cm-1 bands in forms I and II, respectively. Furthermore, note how very different the band structures (peak positions and relative intensities) of the two forms are in the 1200 to 1300 cm-1 region. In fact, most of the corresponding bands in the spectra of the paracetamol forms I and II have different positions to a greater or lesser degree in the region from 1100 to 1700 cm-1. The exception is the band at 1169 cm-1 which is the same for both forms. And so, with adequate spectral resolution, one can readily differentiate paracetamol forms I and II based upon the peak positions of high frequency internal crystal lattice vibrational modes. In general, the polymorphs of molecular crystals can be differentiated even at the high frequencies of the fingerprint spectral region if the Raman spectrometer has a spectral resolution of 1 cm-1 or better.


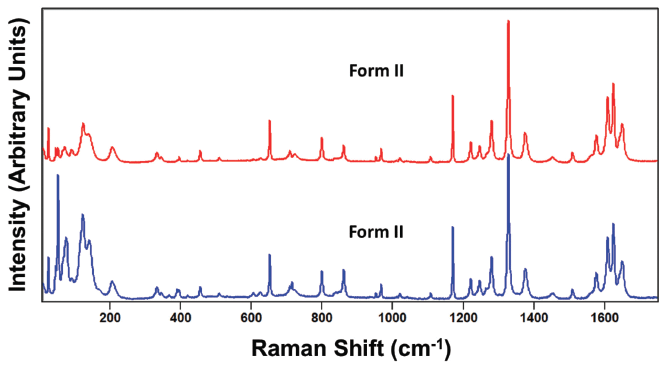
Figure 6: Raman spectra obtained from different grains of paracetamol form II.
The trend observed in the high frequency fingerprint spectral region is observed even in the region from 300 to 700 cm-1, as shown in Figure 8. Keep in mind that the bands appearing in this middle frequency fingerprint region also arise from internal crystal lattice vibrational modes just as do the bands observed in Figure 7. The band structures are even more similar in this middle frequency portion of the fingerprint spectral region. However, if one were restricted to only this segment of the spectrum, the corresponding bands at 466 and 454 cm-1 could quite readily be used to differentiate paracetamol forms I and II, respectively. You may have heard that the lower the frequency of the crystal lattice vibrational mode is the more sensitive it will be to crystal structure. Whereas that statement is generally true when applied to a comparison of external and internal crystal lattice vibrational modes as a group, it is not necessarily true when applied within those two classifications, as we can clearly see from high and middle frequency portions of the fingerprint region shown in Figures 7 and 8, respectively. The lesson to be learned here is that the difference in peak position with respect to crystal form is dependent upon how the different crystal structures affect the molecular interactions within the crystallographic unit cell and thereby determine the energies of the crystal lattice vibrational modes. If there is little difference between the unit cell structures of two crystal forms, then the differences in the energies of their phonon modes and corresponding Raman peak positions will also be very small. Therefore, the ability to differentiate molecular crystal forms based upon the peak positions of internal crystal lattice vibrational modes will be determined by the spectral resolution of the Raman spectrometer.


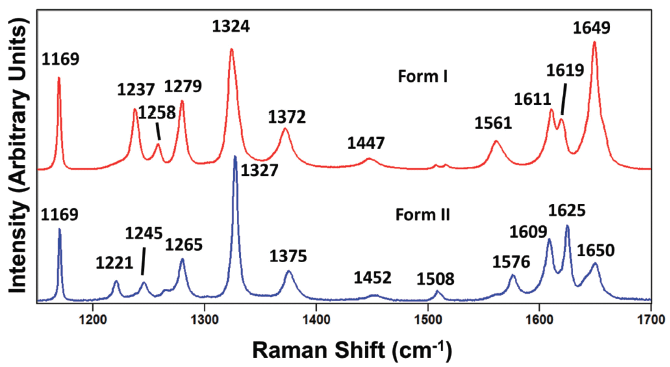
Figure 7: Raman spectra obtained from grains of paracetamol form I (red) and form II (blue).
Turning now to an evaluation of the external crystal lattice vibrational modes, we examine the spectra of paracetamol forms I and II in the region from 10 to 250 cm-1 shown in Figure 9. Here we see significant differences in the peak positions and relative intensities of bands from these two crystal forms. None of the bands in this spectral region appear to have counterparts at the same Raman shift in the other crystal form spectrum; the band structures appear almost entirely different. However, remember that it is not primarily the relative intensities of the bands that are to be relied upon to differentiate the crystals forms but their peak positions due to the energies of the external crystal lattice vibrational modes. The differences in the relative intensities of the Raman bands in the low frequency portion of the spectra acquired from different grains of the same crystal forms shown in Figures 5 and 6 make clear that different relative signal strengths cannot be relied upon to differentiate polymorphs. Rather, the energies of the external crystal lattice vibrational modes and not their relative signal strengths differentiate the crystal forms. Even small differences in the configuration of molecules within the crystallographic unit cell can significantly affect the energies of external crystal lattice vibrational modes. The spectra in Figure 9 very clearly demonstrate the oft heard claim that the low frequency region of the Raman spectrum attributable to external crystal lattice vibrational modes is the most sensitive to crystal structure and thereby the most effective for differentiating multiple forms of molecular crystals. Finally, it is important to note how strong the Raman scattering is from external crystal lattice vibrational modes. In all but one of the spectra shown in Figures 4–6, the signal strengths of the bands below 200 cm-1 are comparable to or greater than the most intense Raman bands in the higher frequency portion of the fingerprint region. Consequently, analyzing the low frequency portion of the Raman spectrum for characterizing polymorphism is particularly advantageous, because it is both sensitive to crystal structure and yields very strong signals.


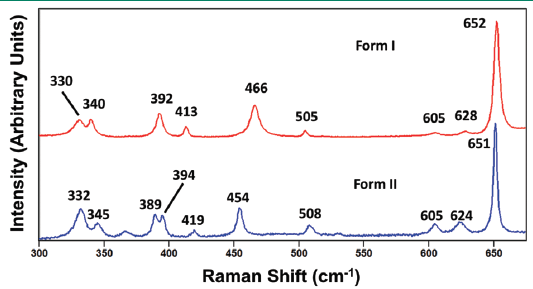
Figure 8: Raman spectra obtained from grains of paracetamol form I (red) and form II (blue).
Carbamazepine Forms I and III
Carbamazepine is a pharmaceutical compound and molecular crystal that can be found in multiple crystallographic forms. Like paracetamol, carbamazepine can be melted or dissolved in a suitable solvent to generate individual molecules. Therefore, the Raman spectrum of solid state carbamazepine consists of bands arising from external and internal crystal lattice vibrational modes as seen in the spectra of carbamazepine forms I and III shown in Figure 10. The spectra of forms I and III appear similar, but are not identical as one might expect from different forms of the same compound. However, just as we saw for the spectra of paracetamol, relative band strengths are not to be relied upon for differentiating crystal forms of the same compound. The spectra shown in Figure 11 were obtained from different grains of carbamazepine form III and yet appear significantly different particularly in the region below 200 cm-1. The band structures arising from both the external and internal crystal lattice vibrational modes appear sufficiently different such that one might incorrectly conclude from the spectra that the carbamazepine samples were of different crystal forms. However, a careful examination of the Figure 11 spectra reveals that where corresponding bands appear in both spectra they all do so at the same peak position to within 1 cm-1, thereby confirming that the grains are of the same crystal form.


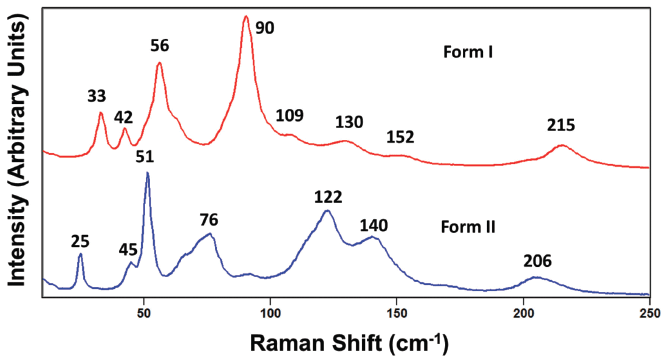
Figure 9: Raman spectra obtained from grains of paracetamol form I (red) and form II (blue).
Carbamazepine polymorph differentiation through Raman spectroscopy requires the acquisition of spectra with resolution sufficient to detect the small differences in peak positions of the various crystallographic forms. Presentation of the carbamazepine forms I and III spectra to show only the 1200 to 1750 cm-1 region in Figure 12 reveals small differences in the peak positions just as we had observed for the different forms of paracetamol. The general patterns of band location and relative intensities are similar. However, all of the peak positions of corresponding bands of forms I and III differ by varying degrees. There are no corresponding bands at the same peak position. The smallest difference is between the 1488 and 1490 cm-1 bands of forms I and III, respectively. Therefore, the high frequency portion of the fingerprint region can be used to differentiate carbamazepine forms I and III.


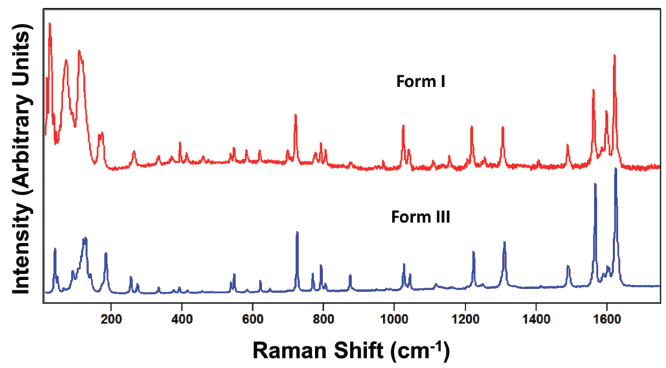
Figure 10: Raman spectra obtained from grains of carbamazepine form I (red) and form III (blue).
Examination of the spectral region between 500 and 1100 cm-1 reveals that most of the corresponding Raman bands of carbamazepine forms I and III have different peak positions that will allow a spectrometer with sufficient spectral resolution to resolve them and differentiate the crystal forms (See Figure 13). It is worth noting here that one can and should use the peak positions and magnitudes of the differences of at least several bands to differentiate crystal forms. For example, the spectrum of carbamazepine form I has peaks at 619, 719, 1025, and 1040 cm-1 for which the corresponding Raman bands in form III appear at 621, 724, 1025, and 1043 cm-1, respectively. Therefore, the magnitudes of the form I minus form III band positions of this set are -2, -5, 0, and -3 cm-1, respectively. Obviously, the more differences in Raman band peak positions that are measured, the more thorough the characterization of or screening for polymorphs will be. Of course, one could simply compare the set of all peak positions to those of the reference spectra of crystal forms that one has already acquired to confirm the crystallographic identity of the sample.


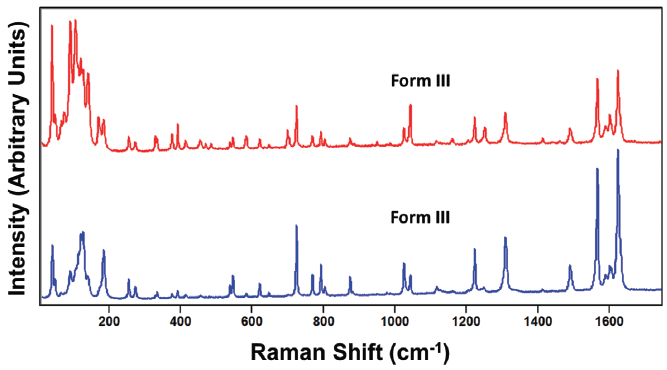
Figure 11: Raman spectra obtained from different grains of carbamazepine form III.
Isn't the measurement of the Raman band differences redundant to just matching the peak positions to reference spectra? Not necessarily, particularly if one encounters a spectrum that doesn't exactly match any of the reference spectra. If the complete set of peak positions of a sample spectrum does not match any of those of your reference spectra, you may have discovered a new polymorph. This is where the measurements of the differences in peak positions can help you characterize this new crystal form. It is likely that the reference crystal form that yields the smallest differences in Raman peak positions relative to those of your unknown or sample spectrum has a crystal structure very close to that of the newly discovered polymorph.


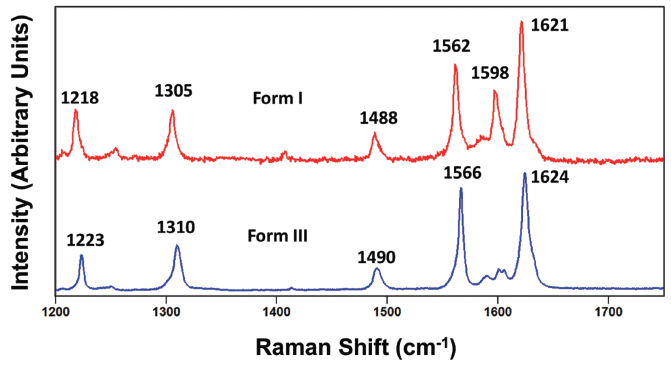
Figure 12: Raman spectra obtained from grains of carbamazepine form I (red) and form III (blue).
Finally, we again review the spectral region between 10 and 300 cm-1 corresponding to the external crystal lattice vibrations. The low frequency portions of the spectra of carbamazepine forms I and III shown in Figure 14 are strikingly different. In fact, it is difficult to even recognize any corresponding bands of the two forms below 200 cm-1 even though a correspondence is clearly recognizable for all of the bands greater than 500 cm-1 throughout the spectral fingerprint region as seen in Figures 12 and 13. Once again, we see how significantly different the peak positions of bands arising from external crystal lattice vibrations are, thereby demonstrating their sensitivity to the unit cell crystal structure and the configurations of the molecules therein. The low frequency portion of the Raman spectrum is clearly the most efficacious for characterizing, differentiating, and screening polymorphs by Raman spectroscopy.


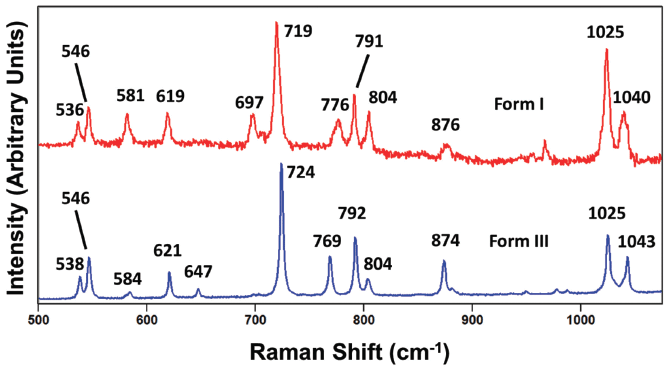
Figure 13: Raman spectra obtained from grains of carbamazepine form I (red) and form III (blue).
Conclusion
Raman spectroscopy is sensitive to crystal structure, and has therefore been successfully used for the characterization and screening of crystal polymorphs. Group theory in conjunction with Raman polarization selection rules has been applied to differentiate the anatase and rutile TiO2 crystal structures by their Raman spectra. Nevertheless, the interpretation of even simple spectra such as those of anatase and rutile TiO2 is not straightforward, and we are aided in our band assignments and spectral interpretation by previously published work. The Raman spectra of molecular crystals were explained in terms of external and internal crystal lattice vibrational modes occurring in the low and high frequency regions, respectively. The energies of the crystal lattice vibrational modes depend upon the configuration of the molecules within the crystallographic unit cell and in particular their molecular interactions. Crystal forms of paracetamol and carbamazepine were differentiated by their peak positions in both low and high frequency regions. Spectral resolution of 1 cm-1 or better is required for effective polymorph differentiation. The low frequency region is the most efficacious for differentiating crystal forms. That is because the energies of external crystal lattice vibrational modes are most sensitive to the configurations of molecules within the unit cell and their molecular interactions. The results very clearly demonstrate the oft heard claim that the low frequency region of the Raman spectrum attributable to external crystal lattice vibrational modes is the most sensitive to crystal structure and thereby the most effective for differentiating multiple forms of molecular crystals. Consequently, analyzing the low frequency region of the Raman spectrum for characterizing polymorphism is particularly advantageous because it is both sensitive to crystal structure and yields very strong signals.


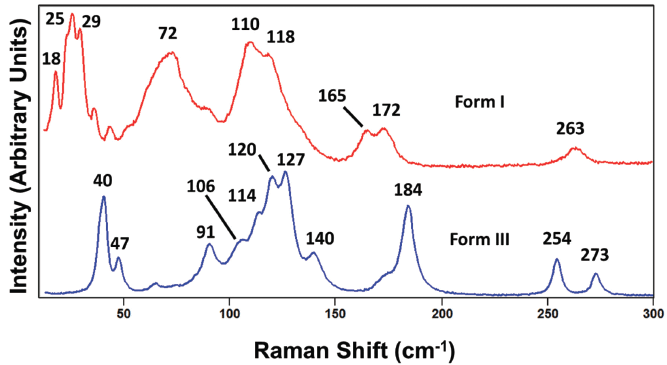
Figure 14: Raman spectra obtained from grains of carbamazepine form I (red) and form III (blue).
References
(1) D. Tuschel, Spectroscopy29(9), 14–21 (2014).
(2) P.M.A. Sherwood, Vibrational Spectroscopy of Solids (Cambridge University Press, London, 1972), p. 4.
(3) D. Tuschel, Spectroscopy30(12), 17–22 (2015).
(4) W.G. Fateley, F.R. Dollish, N.T. McDevitt and F.F. Bentley, Infrared and Raman Selection Rules for Molecular and Lattice Vibrations: The Correlation Method (Wiley-Interscience, New York, 1972), pp. 6–19.
(5) W.G. Fateley, F.R. Dollish, N.T. McDevitt and F.F. Bentley, Infrared and Raman Selection Rules for Molecular and Lattice Vibrations: The Correlation Method (Wiley-Interscience, New York, 1972), p. 165.
(6) G.A. Tompsett, G.A. Bowmaker, R.P. Cooney, J.B. Metson, K.A. Rodgers and J.M. Seakins, J. Raman Spectroscop.26, 57–62 (1995).
David Tuschel is a Raman Applications Scientist at Horiba Scientific, in Piscataway, New Jersey, where he works with Fran Adar. David is sharing authorship of this column with Fran. He can be reached at: SpectroscopyEdit@UBM.com



David Tuschel






A Proposal for the Origin of the Near-Ubiquitous Fluorescence in Raman Spectra
February 14th 2025In this column, I describe what I believe may be the origin of this fluorescence emission and support my conjecture with some measurements of polycyclic aromatic hydrocarbons (PAHs). Understanding the origin of these interfering backgrounds may enable you to design experiments with less interference, avoid the laser illuminations that make things worse, or both.

Raman Microscopy for Characterizing Defects in SiC
January 2nd 2025Because there is a different Raman signature for each of the polymorphs as well as the contaminants, Raman microscopy is an ideal tool for analyzing the structure of these materials as well as identifying possible contaminants that would also interfere with performance.





