Rhenium Coil Field Sampling for Determinations by ICP-AES with Electrothermal Vaporization Introduction
The authors have been characterizing an electrothermal, near-torch vaporization (NTV) micro- and nano-sample introduction system for inductively coupled plasma (ICP) spectrometry.
The authors have been characterizing an electrothermal, near-torch vaporization (NTV) micro- and nano-sample introduction system for inductively coupled plasma (ICP) spectrometry. In this article they demonstrate that portable coiled-filament assemblies can be used for taking part of the laboratory to the sample and concentration determination in a laboratory using NTV-ICP–atomic emission spectroscopy (AES).
Typical inductively coupled plasma–atomic emission spectrometry (ICP-AES) systems have been developed and characterized, and they are frequently used with a pneumatic nebulizer for the introduction of liquid samples into the ICP. Although solid samples are usually acid-digested to a liquid before their introduction into an ICP (also typically using a nebulizer), the focus of this report is on naturally occurring aqueous samples. But before such samples can be introduced into an ICP, they must first be collected in the field.
Conceptually, field sampling involves on-site collection of anywhere between 100 mL to 1 L of various samples followed by their shipment to a laboratory for analysis. There are costs associated with sample transport because of the relatively large volume (and weight) of samples that must be transported from a sampling site in the field to a laboratory for analysis by ICP spectrometry. Extra costs can be incurred if sample storage (sometimes in a cooled or refrigerated unit) is also required before analysis.
Practically, in addition to the costs involved and depending on the analyte of interest, there are chemistry-related issues that must be considered as well. For instance, aqueous samples most likely must be filtered (ideally on-site), typically using a coarse filter. They also must be stabilized (often using a strong acid) before transportation. Sample stabilization is required to minimize analyte loss resulting primarily from adsorption on the walls of the sample container or to prevent precipitation. Safety precautions should also be considered when dealing with the transportation of strong or corrosive reagents to the field (for sample stabilization). Field-stabilized samples are then shipped to a laboratory and are usually stored before analysis.
The elapsed time interval between sample collection and analysis is often critical, especially if there is natural species interconversion — that is, if the species of interest is labile. Let's consider chromium (Cr) as an example: A Cr6+ to Cr3+ interconversion over time has been reported (1). Furthermore, depending on the analyte of interest, acidification may also change the oxidation state of the species of interest. Oxidation-state information (that is, speciation) is important because toxicity is often oxidation-state dependent. Again, chromium is a typical case here: Cr3+ is an essential micronutrient, but Cr6+ is reported as carcinogenic (1). In addition, redox equilibrium is, among others, a function of the final pH of an acidified aqueous sample, the oxygen concentration, and the final ionic strength of the sample. And, it depends on the amount of colloidal and fine particulate material that may still be present in a sample (even after coarse filtering). As already alluded to, the type of acid used to stabilize the sample and the final pH of an acidified sample may shift naturally existing equilibria. It may also help to release particle-bound species or dissociate ligand-bound species (likely both), thus altering the naturally occurring species distribution in the sample. In natural waters, for instance, acidification has been reported to induce a reduction of Cr6+ to Cr3+ (2). Because such chemical transformations begin to take place immediately after sampling, species concentrations determined in a laboratory (after unavoidable delays because of transport and possibly storage) may not necessarily reflect actual species concentration in a sample at the time and conditions (for example, temperature) of sampling. Thus, a species concentration "bias" will likely develop. The United States Environmental Protection Agency (US EPA) recommended that unless stabilized with reagents, Cr-containing water samples "must be analyzed within 24 hours of collection" (2). Additional sample handling steps are required if longer transport and storage times are necessary.
Two conclusions can be drawn from the discussion above. First, elemental analysis can be made cheaper if smaller amounts of sample are brought to the laboratory for analysis. Second, concentration information will more accurately reflect species distribution at the time and conditions of sampling if species are collected separately at their naturally occurring oxidation state and are stored by deposition on an appropriate support. Because it is difficult to bring an ICPA-ES system to the field for on-site measurements, concentration determination of the species deposited in the field must be done in a laboratory.
This article describes an approach that uses removable and interchangeable coiled filament assemblies onto which samples are deposited (for example, by pipetting or electrodepositing samples) on-site. The coils with dried sample residues are subsequently transported to a laboratory where they are used with an electrothermal, near-torch vaporization (NTV) sample introduction system and ICP-AES (1).
Electrothermal, Near-Torch Vaporization Sample Introduction for ICP Spectrometry
A schematic illustration of the instrumentation used for this work is shown in Figure 1a and a photograph of the vaporization chamber and the portable coiled-filament assembly is shown in Figure 1b. The relatively small volume vaporization chamber of the NTV sample introduction system directly replaces the spray chamber of a typical pneumatic nebulizer. In essence, NTV can clip onto any ICP torch with a standard ball joint (Figures 1a and 1b). A coil made from rhenium (Re) filament serves as the sample holder (Figure 1c). The coils were fabricated by manually winding a rhenium filament around a screw with an appropriate diameter. Of the possible elements with high melting points that could be used to make filaments (Table I), Re was selected because unlike tungsten (W) (5,6), Re does not have a known ductile temperature. Thus, unlike W (7), Re does not become brittle after several heating (and cooling) cycles.


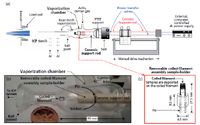
Figure 1: (a) Schematic illustration of the near-torch vaporization (NTV) sample introduction system developed for use with removable, interchangeable, and portable rhenium coiled filament assemblies and micro- or nano-volume samples. (b) Photograph of the vaporization chamber showing the removable coiled filament assembly and the ceramic support rod (coin included for size). (c) Detail of the removable and interchangeable coiled filament assembly. (Adapted with permission from reference 3.)
Unlike previous work with fixed coils and cups (3) or other sample holders of the type encountered in traditional electrothermal vaporization (ETV) sample introduction for ICP spectrometry (5,6), the small coiled-filament assemblies were fabricated to be removable (Figures 1c and 2a). These removable coiled-filament assemblies, with a total mass of 1.4 g each, had identical coupling mechanisms and dimensions so that they could be used interchangeably. Furthermore, removable and interchangeable assemblies with different coil sizes were fabricated, so that volumes ranging from <100 nL to 100 μL could be accommodated. A photograph of the removable and interchangeable coiled-filament assemblies is shown in Figure 2b.


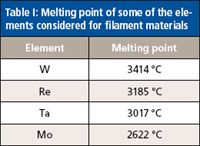
Table I: Melting point of some of the elements considered for filament materials
To interchange coiled-filament assemblies (or to simply deposit a sample by pipetting a micro- or nano-sample into the loops of the coil, Figure 2), the ceramic supporting the coiled-filament assembly was retracted from the vaporization chamber (Figures 1a and 1b). If required, a coiled-filament assembly already installed on the ceramic support was manually removed and a new assembly (for example, of the type shown in Figure 2b) was interchanged on the ceramic. This operation was completed in a few seconds. The ability to easily and rapidly interchange coiled-filament assemblies proved to be beneficial, as we'll discuss later. It is worth noting that for sample pipetting or for coil interchange, the ICP torch operated continuously without interruption. In other words, the ICP was not extinguished.



Figure 2: (a) Removable coiled filament assembly sample holder as installed on the ceramic support rod. For clarity, a red-color food-additive was added and it acted as the sample. (b) Photograph of interchangeable coiled filament assemblies with different capacities shown on a substrate support (coin included for size). (i) coil with capacity â¤1 μL; (ii) coil with 1â10 μL volume capacity; and (iii) coil with 10â100 μL capacity. (Adapted with permission from reference 3.)
The sample introduction sequence into an ICP was as follows: The ceramic support rod with an interchangeable coiled-filament assembly (Figures 1c and 2) attached to one of its ends was removed from the vaporization chamber. A micro- or nano-volume sample was pipetted on the coil and the coiled-filament assembly was reinserted into the vaporization chamber. A seal was formed at the end of the vaporization chamber. Cables running through the ceramic support connected the coiled filament to an external power supply (Figure 1a). The sample on the coil (Figure 2a) was then dried by applying low electrical power from the external power supply (for example, ~100 °C corresponding to ~1 W) to the coil. After drying, the residue that remained on the coil was vaporized into an ICP by applying higher electrical power to the coil (for example, 45 W, corresponding to 2610 °C, as measured using an optical, infrared [IR] thermometer). The vaporized, dried sample residue that remained on the coil was carried to the ICP by an Ar-H2 (for example, 97% Ar, 3% H2) carrier gas. Hydrogen was used to prevent oxidation of the Re coil (3).
Some Analytical Performance Characteristics of NTV-ICP-AES
To provide reference points for comparison purposes, the following brief narrative provides some analytical performance characteristics of the NTV-ICP-AES system. Examples of typical analytical signals are shown in Figure 3a, and their reproducibility is shown in Figure 3b. The detection limits obtained using NTV sample introduction and an ICP optical emission spectrometer equipped with photomultiplier tube (PMT) detectors are included in Table II. The improved precision obtained by NTV-ICP-AES compared to other electrothermal vaporization–based approaches (5,6) is noteworthy.



Figure 3: (a) Typical analyte signals and (b) typical precision.
Taking Part of the Laboratory to the Sample
Instead of depositing samples on a coil by pipetting them in the laboratory (3), samples can be pipetted on removable coiled-filament assemblies (of the type shown in Figures 1c and 2) in the field. Samples on coils can also be dried in the field. Thus, portable coiled-filament assemblies with dried sample residues on them can be shipped to the laboratory rather than shipping large volumes of sample to a laboratory, which reduces shipping, handling, and storage costs.


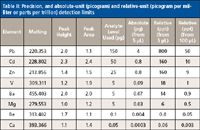
Table II: Precision, and absolute-unit (picogram) and relative-unit (picogram per milliter or parts per trillion) detection limits
Furthermore, instead of pipetting samples on coiled-filament assemblies, samples can be electrodeposited on them followed by a drying step on-site. Electrodeposition also offers added benefits such as preconcentration and matrix cleanup, which can also be performed on-site. Thus, coiled-filament assemblies with matrix-free, preconcentrated dried residues of samples can be shipped to a laboratory where concentrations can be determined by NTV-ICP-AES. In this work, such capability is demonstrated using a single electrodeposition potential and lead (Pb) as the test analyte (4).
By electrodepositing analytes at two distinct potentials (and using different coiled-filament assemblies), different species of the same element can be electrodeposited on coils in the field. Chromium is a typical case where Cr6+ can be electrodeposited at -0.3 V (versus Ag/AgCl) and both Cr3+ plus Cr6+ can be electrodeposited at -1.6 V (versus Ag/AgCl). Shipping two coiled-filament assemblies to the laboratory, one loaded with a dried residue electrodeposited at -0.3 V and both Cr3+ and Cr6+ at -1.6 V allowed Cr speciation in the laboratory by NTV-ICP-AES (1).
It is worth emphasizing that the simplicity of an electrodeposition setup (that is, a sample container, electrodes, and a handheld potentiostat) would make the instrumentation lightweight, small, and portable, and thus suitable for field use.
On-Site Sample Deposition on Coiled Filaments and In-Laboratory Analysis
Previous work has shown that coils with different capacities (ranging from 100 nL to 100 μL as shown in Figure 2b) can be used interchangeably. An example of the use of nanoliter volumes is shown in Figure 4. Subsequently, it was shown that emission signals obtained using interchangeable coiled-filament assemblies with different capacities could be used for constructing calibration curves (3). These results indicate that pipetting different volumes of sample on coils with different volume capacities can be substituted for sample dilution. Although the approach described above was demonstrated in the laboratory, it opens up the possibility for shipping coiled-filament assemblies with dried sample residues on them to the laboratory, thus reducing shipping, handling, and storage costs.



Figure 4: Example signal obtained using 100 nL of diluted Pb standard.
But would dried residues of samples pipetted on coiled filaments and dried on-site remain on the coil during transport, handling, and possibly storage before analysis by NTV-ICP-AES in the laboratory? This question was addressed by placing loaded coil assemblies in a dustproof box, simulating transport by driving the box around, and storing the box in the laboratory for several days before analysis by an NTV-ICP-AES system (Figure 1a). From the results obtained it was concluded that there was no sample loss during transport and storage. Why was this the case?
In the absence of specific adsorption sites, when a metal such as Re and an aqueous sample come into contact, "a metallic surface in contact with water forms a thin layer of a (hydrous) oxide" (8), most probably a monolayer (1). Such monolayers are thermodynamically favorable because they reduce the surface free energy; in fact, such layers are subject to the same equilibria as bulk metal oxides (8). Based on these conclusions, it can be speculated that -Re-OH2+, -Re-OH, or -Re-O- is formed. If a metal ion is also present in the sample (and depending on the pH of the sample, the potential of zero charge of the Re surface, and ignoring multivalent ions), it can be speculated that the following reaction takes place (1):


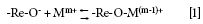
From this reaction, it can be concluded that cations are attached to the surface by a phenomenon characterized by a strong binding energy (such as chemisorption) rather than one with comparatively weaker binding energy (such as physisorption). In fact, it has been reported that in some cases chemisorbed species on surfaces require strong ion exchangers (such as acids) to be eluted from a surface they are attached to (1). Alternatively, the application of high temperature is required to vaporize chemisorpted species (as is the case here). It is worth noting that chemisorption on metal surfaces in contact with aqueous solutions is a common phenomenon (9–12), and that it occurs spontaneously and with rapid kinetics (10). Based on the above, and because of the bond formation between Re and Mm+ (as suggested by equation 1), it is not surprising that dried solution residues remain on the coil during transport and storage.
These ideas were confirmed by determining copper (Cu) in tap water. Copper in tap water originates from plumbing materials. Tap water (obtained from a local tap) was diluted on-site using 18.2-MΩ·cm water and was air dried on-site. Coiled filaments with dried, Cu-containing residues on them were then transported to the laboratory where Cu concentration was determined using NTV-ICP-AES. The Cu concentration was found to be below the action level required to trigger the EPA's lead–copper rule (13). The rule stipulates that "if Pb concentration exceeds an action level of 15 ppb or if the Cu concentration exceeds an action level of 1.3 ppm in more than 10% of the customers taps sampled," some kind of corrective action must be taken. Thus far, to test for this rule, only Cu has been dealt with. Lead concentration determination is described below.
On-Site Pb Electrodeposition on Coiled Filaments
Rather than simply pipetting samples on coiled filament assemblies, is it possible to electrodeposit analytes on such filaments? In addition to preconcentration, electrodeposition offers some analyte selectivity and provides preconcentration and matrix cleanup. Pb was selected as the analyte of choice because of its environmental significance and because it has one of the poorest detection limits by ICP-AES. The experimental set up for Pb electrodeposition is shown in Figure 5. A coiled-filament assembly served as the cathode and a platinum (Pt) wire was used as the anode. To simplify instrumentation and further enhance portability, a reference electrode was not used. To facilitate on-site operation, a homemade, handheld, battery-operated power supply was used. The interelectrode distance was kept constant, the sample volume was arbitrarily selected to be 20 mL, and the electrodeposition time was 3 min. Because of its simplicity and light weight, the electrodeposition instrumentation was portable.


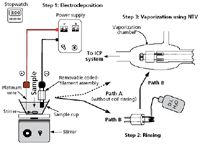
Figure 5: Experimental set up and sample processing steps used for Pb electrodeposition. (Adapted with permission from reference 4.) See text for discussion.
For electrodeposition, metal ions such as Pb2+ can be deposited anodically or cathodically (4). In this case, cathodic deposition (by reducing Mn+ to M0, according to equation 2) was selected.

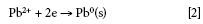
It has been reported that Pb2+ (because of its high hydrogen over-potential), can be electrodeposited from acidic solutions with efficiency close to 100%. To facilitate portability and because of the minor effect of temperature (4), the effect of temperature was not considered.
Applicability of Pb electrodeposition on coiled-filament assemblies in the field and analysis in the laboratory by NTV-ICP-AES was demonstrated using the determination of Pb in hard tap water. Typical hard water has a mineral content greater than 180 mg/mL (expressed in CaCO3 equivalent concentration [14]). The water sample was taken from a local residential tap that was fed with one of the "hardest" groundwaters in Canada (15) (with water hardness reported to be as high as 960 mg/L in CaCO3 equivalent concentration [15]). For comparison, soft waters have a hardness of <60 mg/L (16).
In many parts of the world, Pb in drinking water comes from aging Pb water pipes as well as from solder and flux used for plumbing purposes. According to the US EPA (13), an action level for Pb in tap water is set at 15 ng/mL. Guidelines for Canada and those published by the World Health Organization (WHO) (17) are set at 10 ng/mL (18).
The approach described above was tested in the field by placing the instrumentation shown in Figure 5 in a Plexiglas enclosure (thus preventing contamination from airborne particulate matter). Subsequently, Pb electrodeposition using tap water took place for 3 min. After electrodeposition and air-drying, a salt deposit was visibly noticeable on the coil. When the dried electrodeposited sample including the salt deposit were vaporized into an ICP (as described by path A in Figure 5), a bright yellow-orange emission was visually noticeable. This intense yellow-orange emission caused stray light and a double peak (for example, one for Pb and the other from stray light) at the Pb wavelength, with the peak caused by stray light interfering with the Pb emission signal. To eliminate the peak because of stray light, a rinsing step with 18-MΩ·cm water was added following electrodeposition (as shown in path B in Figure 5), thus rinsing away the salt deposit. This step eliminated the peak caused by stray light. Rinsing did not cause any noticeable Pb loss from the coil because during cathodic deposition a metal ion is reduced to the metallic form (equation 2). As discussed earlier, it can be suggested that Pb was chemisorbed on the coil, thus with-standing mechanical vibrations during transportation. Additional evidence has been provided by others who also reported the use of electrode rinsing (4). In one case, it was reported that electrode rinsing is a "standard practice" (4). The portable coiled-filament assemblies with Pb electrodeposited on them were rinsed and dried on-site, and were subsequently transported to the laboratory where the Pb concentration was determined by NTV-ICP-AES. The Pb concentration was found to be below the action level for Pb set by the EPA's lead-copper rule (13). Additional details such as validation and precision information can be found in reference 4. An example signal obtained after Pb electrodeposition is shown in Figure 6. The signal shown in Figure 6 is in the parts-per-trillion (pictogram per milliliter) level, which is clearly below the current detection limit of ICP-AES with pneumatic nebulization sample introduction. The Pb detection limit following a 3-min electrodeposition was 25 pg/mL (or 25 ppt); according to the data included in Table II, this limit is >30 times better than that obtained without electrodeposition from 5-μL samples.

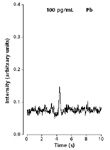
Figure 6: Example signal obtained after electrodeposition of Pb. See text for discussion.
As briefly described earlier, the Cu concentration from the same tap water was determined by direct pipetting on coiled-filament assemblies. Thus, direct pipetting of Cu and electrodeposition of Pb on portable coiled filaments can be used for compliance testing of the EPA's lead-copper rule.
On-Site Electrodeposition for Cr Speciation
Because of the success of the electrodeposition approach described above for Pb, the following question arose: Is it possible to perform controlled-potential electrodeposition using two different coiled-filament assemblies and two distinct electrode potentials to deposit two different species of the same element? In other words, can electrodeposition be used for Cr speciation?
The electrochemical cell used for Cr speciation is shown in Figure 7. It consists of a polyethylene beaker and a removable cap that supports the electrodes (1). The cap was supported by an adjustable arm, thus also controlling the immersion depth of the electrodes. The coil served as the working electrode and a coiled platinum wire served as the counter electrode. A compact Ag/AgCl electrode was used as the reference electrode. The coils were preconditioned by applying high electrical power to them (for example, 45 W corresponding to 2610 °C) for 5 s before their use. For selective electrodeposition of Cr species, a voltage of -0.3 V (versus Ag/AgCl) was used for electrodeposition of Cr6+ and a voltage of -1.6 V was used for electrodeposition of both Cr6+ and Cr3+. The Cr3+ concentration was determined by difference. At the end of a 60-s electrodeposition time, the cell cap was raised, the coiled filament assembly was removed from its holder in the cup, and the assembly was carefully rinsed with distilled, deionized water (18 MΩ), thus removing any salt deposit on the coil.


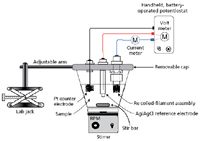
Figure 7: Experimental setup used for Cr speciation by electrodeposition.
For method development, calibration, and validation, artificial seawater was used (1). An acetate buffer was added immediately before electrodeposition (to buffer the sample to pH 4.7). The buffered artificial seawater was immediately spiked with known amounts of Cr3+ and Cr6+, and the spiked solutions were used right away. Chromium emission signals from buffer blanks were negligible. Natural surface seawater samples were collected off-shore from Halifax Harbor (Nova Scotia, Canada). Then, they were filtered through a prewashed 0.45-μm filter and were immediately buffered to pH 4.7 with 15 mL of the buffered sample placed into a PTFE beaker that served as the electrochemical cell (Figure 7). Chromium species in seawater were immediately electrodeposited for 60 s on coiled-filament assemblies. The coils were washed with deionized water, air dried, and subsequently transported to the laboratory where the Cr species were determined by NTV-ICP-AES. The Cr species concentrations were found to be well below the maximum levels set by the EPA for saltwater and continuous exposure.
The emission signals shown in Figure 8 have been included for indirect (for example, Cr3+, Cr6+ versus Cr) comparisons of the signal enhancements obtained because of 60 s of electrodeposition. For more-direct comparisons, the detection limit (3σ) for Cr was found to be 0.5 ng/mL (or 2.5 pg in absolute amount). This detection limit is 200-fold superior to that reported for Cr using graphite-based ETV-ICP-AES without the use of chemical modifiers and ~sixfold better than ETV-based methods using chemical modification (19). When gains because of electrodeposition are factored in, rough improvements range from 250- to almost 13,000-fold (1). Additional details (such as validation and precision) can be found in reference 1. These results compare favorably with those reported by others for Cr speciation of river water using microcolumns (20).



Figure 8: (a) and (b) Cr signals obtained after a 60-s electrodeposition; (c) Cr signal obtained without electrodeposition. The differences in concentration between (a), (b), and (c) are noteworthy (Adapted with permission from reference 1.)
Conclusions
Bringing a small part of the laboratory to the sample in the form of small, light-weight, portable, removable, and interchangeable coiled-filament assemblies of a near-torch vaporization micro- and nano-sample introduction system provided unique capabilities, such as improved limits of detection (versus those obtained by pneumatic nebulization sample introduction) and reduced shipping and storage costs, thus reducing cost per analysis.
Going from depositing a sample directly on a coiled filament (for example, by pipetting) to using a coil as one of the electrodes of an electrochemical cell enabled matrix cleanup and preconcentration. Preconcentration enabled the determination of Pb at concentration levels well below the current detection limits of ICP-AES systems equipped with a pneumatic nebulizer for sample introduction. It also enabled the analysis of Cr species, thus facilitating Cr-speciation measurements.
Although the NTV micro- and nano-sample introduction system described here has been used with ICP-AES, the concepts, methods, and approaches are applicable to ICP coupled to mass spectrometry (MS) as well. In the future, simultaneous multielement determinations will be attempted.
References
(1) H.R. Badiei, J. McEnaney, and V. Karanassios, Spectrochim. Acta, Part B 78, 42–49 (2012).
(2) Determination of Hexavalent Chromium in Drinking Water by Ion Chromatography with Post-Column Derivatization and UV-Vis Spectroscopic Setection (US Environmental Protection Agency, Washington, D.C., Method 218.7), November 2011.
(3) H.R. Badiei, B. Lai, and V. Karanassios, Spectrochim. Acta, Part B 77, 19–30 (2012).
(4) H.R. Badiei, C. Liu, and V. Karanassios, Microchem. J. 108, 131–136 (2013).
(5) S.N. Hanna and B.T. Jones, Appl. Spectrosc. Rev. 46, 624–635 (2011).
(6) S.N. Hanna, C.P. Calloway, Jr., J.D. Sanders, R.A. Neslon, J. Cox, and B.T. Jones, Microchem. J. 99, 165–169 (2011).
(7) V. Karanassios and K.P. Bateman, Spectrochim. Acta, Part B 49, 847–865 (1994).
(8) N. Lallay, Z. Torbie, M. Golic, and E. Matjevic, J. Phys. Chem. 95, 7028–7032 (1991).
(9) R.O. James and T.W. Healy, J. Colloid Interface Sci. 40, 42–52 (1979).
(10) M.A. Anderson and A.J. Rubin, Eds., Adsorption of Inorganics at Solid-Liquid interfaces (Ann Arbor Science,1981).
(11) G.D. Parfitt and C.H. Rochester, Eds., Adsorption from Solution at the Solid/Liquid Interface (Academic Press, 1983).
(12) A.A. Zagorodni, Ion Exchange Materials: Properties and Applications (Elsevier, 2007).
(13) Lead and Copper Rule (LCR), and National Primary Drinking Water Regulations for Lead and Copper 40 CFR Parts 9, 141 and 142 (US Environmental Protection Agency, Washington, D.C.), January 12, 2000.
(14) W. McGowan, Water Processing. Residential, Commercial, Light-Industrial. 3rd Edition (Water Quality Association, 2000).
(15) S. Gombos, "Residential water softener performance study," Region of Waterloo, Testing report #1, April 2011.
(16) "Hardness in Drinking Water," (World Health Organization, WHO/HSE/WSH/10.01/10/Rev/1, 2011).
(17) Guidelines for Drinking-Water Quality [electronic resource] Incorporating First Addendum. Vol. 1, Recommendations-3rd ed. (World health Organization, 2006).
(18) Guidelines for Canadian Drinking Water Quality (Health Canada, Dec. 2010.)
(19) W. Fuyi, J. Zucheng, H. Bin, and P. Tianyou, J. Anal. Atom. Spectrom. 14, 1619–1624 (1999).
(20) A.G. Cox and C. McLeod, Microchim. Acta 109, 161–164 (1992).
Hamid R. Badiei, PhD, is with PerkinElmer, Inc., in Woodbridge, Ontario, Canada.
Vassili Karanassios is a professor of chemistry in the Department of Chemistry at the University of Waterloo in Waterloo, Ontario, Canada. Direct correspondence to: vkaranassios@uwaterloo.ca.











Trending on Spectroscopy: The Top Content of 2024
December 30th 2024In 2024, we launched multiple content series, covered major conferences, presented two awards, and continued our monthly Analytically Speaking episodes. Below, you'll find a selection of the most popular content from Spectroscopy over the past year.


Best of the Week: Hyperspectral Imaging, ICP-MS Analysis of Geological Samples, Product Roundup
October 18th 2024Top articles published this week include an article about hyperspectral imaging in human skin research, a peer-reviewed article about analyzing geological samples using atomic spectroscopy techniques, and an equipment roundup piece about the latest products in the industry.

Analyzing Mercury Isotopes: A SciX Interview with Frank Vanhaecke, Part 2
October 17th 2024In this preview to the upcoming SciX 2024 conference next week, Spectroscopy sat down with Frank Vanhaecke to discuss about his educational background and what he is looking forward to about the upcoming conference.

