Transformative Mass Spectrometry
Some central transformative themes and their impact in modern analytical mass spectrometry are discussed, such as isotopic analysis, exact measurements, information processing, and more.



Chronologies sequence significant developments in mass spectrometry (MS), although there is always some imprecision in determining the "proper" date for the conceptualization or adoption of an idea. Because hindsight offers a better focus, significant advances and the publications that describe them are sometimes more easily recognized years after they first appear. But it's not necessarily a singular date that is important. Instead, a clear description and execution of a new approach, and its subsequent adoption, often in an unexpected arena, can result in a transformative development in the practice of MS. This column describes some central transformative themes and their impact in modern analytical MS.
A comprehensive history of mass spectrometry (MS) would delineate, inter alia, the use of MS to identify isotopes, confirm the structure identity of both inorganic and organic samples, support geochemical applications through isotopic analysis, present a flexible platform for mixture analysis specifically with environmental applications, and describe its use in studies delineating fundamental ion reactivity, biomolecular identification and characterization, forensic chemistry, process control, and numerous "-omics" applications. Certainly, there are research and applications areas that have been missed in this list, but such is the nature of MS. Also clearly, this is not a list of sequential developments. Instead, it is a compilation of independent active application venues in which MS plays a supporting, if not a central, role. An overview of these areas from the broadest possible perspective allows underlying themes to be identified. Some of these are common across many applications and can be considered transformative. This column describes those transformative themes and links them to the modern day applications of MS. Figure 1 separates transformative themes into a central group of three and a surrounding cohort. The central themes are isotopic analysis, exact measurements, and information processing.


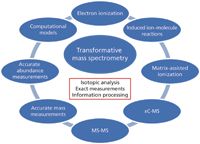
Figure 1: Three central themes (in red) supporting transformative modern mass spectrometry, surrounded by concept areas representing distinct transformative research and experimentation.
Central Themes
Isotopic Analysis
We have discovered all of the stable isotopes and measured their masses and abundances. In MS, the distribution of isotopes in atomic and molecular ions, in fragment ions resulting from dissociations, or in supramolecular ions, can be predicted from natural occurrence and has been mapped as a function of differential reaction rates, or specifically designed artificial manipulation. When we measure the mass of an ion, we use that mass to deduce atomic and empirical formula composition, using our knowledge about the mass and abundance distribution of isotopes. We can also use our knowledge about isotopes to serve as a quality check on our measured data, both in terms of the measured mass and the measured abundance. For example, for a given number of 12C atoms in an ion, there is a known and predictable number of 13C atoms. Ions with distinctive isotopic envelopes (such as chlorine or bromine) are visually evident in a mass spectrum. Isotopic analysis, and our knowledge of isotopic distribution, underlies every facet of modern MS. If the isotopic distribution varies from what we expect, either our measurement is inaccurate or we are looking at the end result of a differential phenomenon.
Exact Measurements
To repeat (with a slightly different emphasis), we have discovered all of the stable isotopes, and we have measured their exact masses and their exact abundances. Those measurements are accurate (true) and precise (reproducible) to extraordinary tolerances. They are "exact," and ongoing research is focused on making them more so. Exact measurements at the limit requires sophisticated and specialized instruments, and "exacting" protocols for sample collection, preparation, and analysis. Measurement performance has steadily improved with each generation of MS instrumentation. Accurate mass measurement has become more accessible across the board. Dynamic ranges are extended with better electronics. As a consequence, instrumental sensitivity increases, and consequently, limits of detection and quantitation trend lower and lower. Better analytical performance places new demands on sample collection and sample storage protocols, with the result that the overall error is often determined by noninstrumental factors.
Information Processing
MS is inherently a multidimensional and data-rich analysis method. Even when light-beam oscillographs were used to record the data, the three concurrent traces (for example, 1×, 10×, and 100×) provided an automated means of scaling intensity measurements. When film was used as the detection method, as in a focal plane spark-source mass spectrometer, a scanning densitometer (and subsequent processing) converted film exposure as a function of plate position into intensity as a function of mass. Digitization of the analog output of a detector allowed for data system recording of data, comparison of data, and archiving and sharing of data. Higher digitization rates and higher-bit transformation meant more and more data. But importantly, after the data was in a digital format, it could be archived, shared, processed with statistical methods, assessed with information theory, and displayed in more meaningful ways. Effective information storage and processing is rapidly becoming a concern across-the-board in modern MS applications.
Surrounding Themes
Surrounding themes incorporate the analytical capabilities derived from each of the central themes described above. Each theme represents a separable and distinctive premise in the use of MS, which the following vignettes distill independently of specific applications. Some transformative developments have been recognized with prizes and honors from the community, and others are overdue for such acknowledgement.
Electron Ionization
Basic MS had been in place for some 30 years, typically used for the discovery of elemental isotopes, before its potential applicability to organic analysis became apparent. Gases were readily introduced into the mass spectrometers of the times and certain elements could be ionized directly from filaments. Other elemental samples had no significant vapor pressure and could not be analyzed by MS until suitable methods of sample introduction and ionization were devised. Such methods were promptly devised. A few organic molecules were sufficiently volatile that their vapors could be swept into the mass spectrometer. As long as their partial pressures exceeded that of residual gases in the system, ions derived from these compounds could be identified. But the processes through which the ions were formed were less clear. Sample introduction systems, such as the direct insertion probe, provided a means to introduce many organic samples as gas-phase molecules into the source of the mass spectrometer. But it was the transformative development of electron ionization as a reproducible and generally applicable ionization method that sparked the revolution. In the electron ionization (EI) source, electrons from the filament interact with the gas-phase molecule to cause the ejection of an electron, leaving an odd-electron molecular ion M+.. Two factors make electron ionization a transformative theme. The first is that the pressure in the EI source as it was ultimately developed was sufficiently low that the creation of the molecular ion and its subsequent dissociations were unimolecular events. Neither the molecule nor the ion created from it collide or interact with any other species. Therefore, all of the fragment ions in the mass spectrum must originate from the molecular ion in simple or successive rational dissociative reactions. Ultimately, these chemical reactions could be interpreted, collected, modeled, and, to an extent, predicted. The second factor is that the ionization itself is a Franck-Condon transition. The removal of the electron is so fast that the molecular structure does not change during ionization. Although dissociations may result in or require rearrangement, the structural starting point is set, and interpretation of the spectrum can evidence that structure.
Induced Ion or Molecule Reactions
Nothing in MS stays simple for long. Electron ionization provided a theme in which we could consider the isolated unimolecular reactions of ions. But there were hints of ion or molecule reactions, such as surface reactions with the metal walls of the EI source. The development of chemical ionization (CI) MS in the mid 1960s was the direct result of a generalized theme of induced ion reactions in MS. In CI, neutral sample molecules react with reagent ions. Such reactions had been studied for years in other venues, such as those concerned with reactions of charged and neutral species present in the atmosphere. The broad impact of CI was that (using methane as the reagent gas, for example) it brought the predictability of acid or base chemistry into the gas phase and into MS. As an ionization method, CI could be energetically tuned. Further, after the idea of a selected reagent ion took hold, all manner of reagent ions could be used, for both positive and negative ion analysis, and with a designed selectivity. The choice of the reagent ion became part of rational selectivity in analysis. The concept that reactions in the rarefied vacuum of the mass spectrometer could be designed and exploited became a theme in MS that continues to the present day. A second part of this theme encompasses the idea that ions, after they are formed, can be prompted to react, again by an induced reaction. Collision-induced dissociation is the classic example of such an activation, and surface-induced and photon-induced dissociation, and more recently electron-capture and electron transfer ion activation methods are also included. Where in the mass spectrometer we choose to make such reactions occur, whether in the ionization source or elsewhere, is linked to several other themes described here.
Matrix-Assisted Ionization
In EI, ionization takes place in an isolated, well-controlled gaseous environment. In CI, the reagent ion is present in great excess of the sample molecules, and so ionization of sample molecules again takes place in a controlled and characterized gaseous mixture. Such environments may seem far preferable to a situation in which a matrix effect might be observed, such as ionization in the presence of a liquid or solid matrix. However, the term "matrix effect" has commonly been burdened with a negative connotation. Matrix effects were those that decreased sensitivity, skewed selectivity, or added noise and uncertainty to the measurement. Central to these deleterious issues was the fact that the composition of the matrix was unknown or not controlled. On the other hand, matrix-assisted ionization methods, including electrospray ionization (ESI), matrix-assisted laser desorption–ionization (MALDI), and their precursors in matrix-assisted secondary ion MS and fast atom bombardment, have as a central tenet the control of the physical and chemical properties of the matrix. The matrix augments and mediates the ionization process. It is worth noting that matrix effects that increase the efficiency of ionization have an even longer history in isotopic geochemistry. In the more modern applications of MALDI and ESI, the matrix is a bridge to assist desorption of sample molecules and ions, provide a route to ionization through protonation or cationization, control the flux and form of ionization energy, and physically separate sample molecules and potentially reactive species. Rather than eschewing a matrix, informed selection of an ionization matrix of known properties became a tool to aid in the analysis. Then, as we became more adept at assessing the mass spectra produced, novel interactions between the same molecules or ions and the matrix could be explored, such as the unfolding of complex biomolecular ions in liquid matrices of various properties.
xC–MS
In its classical application of MS to the identification of compounds, and the discernment of molecular structure, a basic assumption is that one compound is present in the ionization source at any given time. The ions observed in the mass spectrum therefore should all be correlated to that compound, and that structure, through rational fragmentation reactions. Real-world samples are almost always mixtures. Consider an environmental sample; it may contain tens of primary components at similar concentrations, and hundreds of secondary components at 10–100× lower levels. Column chromatography, independently of the use of MS as a detection method, was developed as a means to separate such mixtures. Analytical gas chromatography (GC) was developed first in the 1950s, and liquid chromatography (LC) in the 1970s, followed by other column-based methods (xC). These were transformative developments in and of themselves. Consider that in a benchtop distillation, a physical separation of mixture components is achieved, with separated components collected into separate flasks, for example. In analytical GC, no such physical separation of components occurs. Instead, different sample components require a different transit time through the column; a detector at the column exit provides a signal for each eluted compound. The signal output can provide some information about the component (as in an electron-capture detector contrasted with a thermal conductivity detector). However, the end result of this analysis is not physically separated compounds; instead it is information that characterizes the mixture, much of which may remain undisturbed. Information is the transformative end result. But the use of a mass spectrometer as a detector provides the ultimate unique information signature for each component in the mixture — its mass spectrum. On-line analytical separations combined with MS detection transformed specialized MS into a widely adopted general analytical tool. The informing power of the so-called hyphenated methods, so self-evident now, was a salient topic of research in the early 1980s.
MS-MS
As sector-based mass spectrometers (such as a Nier-Johnson geometry instrument consisting of an electric sector followed by a magnetic sector) were used for organic compound analysis, broad signals were noticed at the spectral baseline. These signals were interspersed with the normal sharp peaks. Ultimately, they were identified as signals arising from metastable ions. Metastable ions are those ions which dissociate in field-free regions (such as between the source and the first sector) as they traverse the mass spectrometer. Passing the ions through a localized region of higher pressure, reversing the order of the sectors, and decoupling the sectors led to the development of mass-analyzed ion kinetic energy spectrometry (MIKES). The transformative factor in MIKES was the espousal of a process that induced purposeful reaction of a mass-analyzed ion as it passed through two independently operated sections of the instrument, and the assessment of the ionic products of that reaction. The same process is exemplified in the triple-quadrupole mass spectrometer; the first quadrupole selects an ion by mass. The ion is activated in the central rf-only quadrupole, and the ionic products of the activated reaction are mass-analyzed by the third, independently operated, quadrupole. By analogy to xC–MS, the first mass analysis is analogous to a separation, here accomplished through separation by mass rather than differential retention on a column. The transformative aspect was the speed with which ions can be selected, activated, and analyzed, and then the selected factors changed, resulted in a multiplexed analytical instrument that could rapidly produce large volumes of data, and could be reconfigured in real time for complex mixture analysis. Hours became minutes, and sometimes seconds and milliseconds. MS-MS could be extended to MSn. With ion-trap instrumentation, the sequence of processes could be extended almost to the limits of ion detection (a few hundred ions), with the flexible use of the MS-MS process as a series of analytical filters designed to preserve signal over noise.
Accurate Mass Measurements
Because we know the exact masses of all the isotopes, and the distribution of these isotopes, we can predict the appearance of the molecular isotopic envelope for a molecular ion of known empirical formula. We can compare the measured envelope to the predicted envelope as an assessment of the measurement quality. Conversely, when confronted with an ion of unknown formula, we can use the measured mass to narrow the realm of possibilities, or to establish the formula with a given degree of certainty. The latter was an early classical application of higher-resolving-power instruments in organic analysis; determination of the ion empirical formula helped to link the ions and their fragmentation reactions. An accurate mass measurement can be used as a difference measurement to establish the formula of the fragment species lost. The usefulness of an accurate mass measurement has evolved, such as its use for tracing the incorporation of stable isotopes or mass defect labels as part of a sample derivatization strategy. In each of these applications, the transformative value of an accurate mass measurement comes from the interplay between necessary and sufficient conditions. Conventional understanding cautioned that as the mass of ions increases, the number of possible formulas increase, and the ability to definitively establish a formula diminishes. But a m/z scale in a mass spectrum has two factors. Increasing the charge "z" on an ion increases the effective mass range of an instrument. But another transformative facet of accurate mass measurement is revealed in the calculation of an accurate molecular mass from repetitive measurements of multiple charged molecular ions formed in electrospray ionization. In these measurements, random errors are mitigated by the greater number of independent measurements, a basic statistical tool with pervasive application in MS.
Accurate Abundance Measurements
With specialized instrumentation, the abundance ratios between certain stable isotopes present in a sample can be determined with astoundingly high accuracy and precision. For example, the Vienna Standard Mean Ocean Water (VSMOW) is a suggested standard for the isotopic composition of freshwater, and is given as 2H/1H = 155.76 ± 0.1 ppm. Variations from this composition, within that stated limit of error, are used to discern differential processes in hydrological systems, predicated on that fact that an exact measurement can be achieved. Other stable environmental isotopes include those of carbon, nitrogen, oxygen, and sulfur. Measurements of these isotopes in various terrestrial samples track the carbon cycle (especially through plant photosynthesis), and can track the passage of nutrients in the biosphere. Sample dating measurements depend on these accurate abundance ratio measurements. These measurements are, of course, not limited to Earth's biosphere, or the terrestrial realm. Isotopic studies in cosmochemistry help to discern some of the earliest fundamental processes, even those that predate the creation of our own solar system. The importance of sample collection and sample provenance in all of these studies should be self-evident.
Computational Models
Reactions of ions such as dissociation to lead to fragment ions can be described in the terms used to describe any other reaction. There is an activation energy related to the observed rate of reaction, a distribution of energy in an activated transition state, and a structure associated with that transition state. Progress along a unimolecular reaction coordinate from reactants to products can be described by energy curves derived from experimental observation, but also from computational models. The calculational model (Rice-Ramsperger-Kassel-Marcus [RRKM]) could be used to produce a theoretical mass spectrum, if the internal energy distribution of the isolated ion was known (1). Calculational models, however, have a much broader impact on MS. Polyatomic ions of moderate structural complexity can be modeled and the probable structure of lowest energy established. Detailed space-filling models can provide insights into reactions such as the folding or unfolding of complex biomolecular structures. On an instrumental platform, expansive improvements in computational performance and data storage have transformed the archival use of data systems in the compilation and searching of mass spectral libraries. Libraries have been supplanted with databases and data mining searches the databases to establish new correlations and patterns in the data. Improved computational capabilities are used to compile and compare the terabytes of data measured in "-omics" research. Computational models are used to track stable isotope incorporation through analysis of the isotopic profile of ions formed from products of metabolic processes, and hydrogen or deuterium exchange reactions, through multiple generations of processes. Simply stated, modern MS research would not be tenable without underlying computational support.
Conclusions
It was with some trepidation that this collection was compiled. The themes are broad enough so that the danger of a blatant omission might be avoided, but the choice of themes is sure to leave some dissatisfied that their field of application or research did not receive adequate attention. Still, on reflection, the centrality of themes, and the organization of the related developments coalesced into the coherent group presented here. If there is to be debate, let it commence. As quoted in Hughes (5), Frederick Soddy wrote: "So easy is it to fall into the error of thinking that things which look obvious after a discovery were just as obvious before." As a science, MS has routinely amazed its adopters, and still often confounds its practitioners, because it evolves so quickly, and spreads rapidly through new and unexpected vectors of application. MS is founded on the simplest and most basic of scientific principles, and yet makes au courant contributions to the most complex modern analytical issues, and reflects the ongoing scientific challenge to reach analytical certainty, as reflected in this quote from Aston's Nobel prize celebration "No probable, possible, shadow of doubt, no possible doubt whatever" (5).
References
(1) J. Griffiths, Anal. Chem. 80, 5678–5683 (2008).
(2) http://masspec.scripps.edu/mshistory/mshistory.php.
(3) A History of European Mass Spectrometry, K.R. Jennings, Ed. (IM Publications, 2012), ISBN: 9781906715045.
(4) T. Baer and P.M. Mayer, J. Amer. Soc. Mass Spectrom. 8, 103–115 (1997).
(5) J. Hughes, Dynamis 29, 131–165 (2009).
Kenneth L. Busch premiered the first "Mass Spectrometry Forum" in February, 1994. Two decades and about 75 columns later, this adventure draws to a conclusion with this last column. Thank you to the readers who have perused the columns over the years, and who have responded with comments and ideas. I am grateful to the editors at Spectroscopy who made this opportunity available. I will someday convince them that it is properly MS/MS and not MS-MS, but the world will survive regardless. This column is the sole responsibility of the author, who can be reached at wyvernassoc@yahoo.com



Kenneth L. Busch







Mass Spectrometry for Forensic Analysis: An Interview with Glen Jackson
November 27th 2024As part of “The Future of Forensic Analysis” content series, Spectroscopy sat down with Glen P. Jackson of West Virginia University to talk about the historical development of mass spectrometry in forensic analysis.

Detecting Cancer Biomarkers in Canines: An Interview with Landulfo Silveira Jr.
November 5th 2024Spectroscopy sat down with Landulfo Silveira Jr. of Universidade Anhembi Morumbi-UAM and Center for Innovation, Technology and Education-CITÉ (São Paulo, Brazil) to talk about his team’s latest research using Raman spectroscopy to detect biomarkers of cancer in canine sera.



