Simple Spectrometer System, Simple Validation?
Computerized System Validation (CSV) is usually associated with great mountains of paper (GMP). Why should this be the case? Ask yourself, am I lazy? Why write multiple documents if only one is required to validate a spectrometer with a simple intended use? Interested? Read on…
Today’s the day you have been waiting for! Your brand-new UV-visible (UV-vis) or infrared (IR) spectrometer and controlling software is due to arrive in your regulated laboratory. The instrument’s location and services are ready. The supplier’s engineer is on hand to install and qualify the instrument against your user requirements. You’ve reviewed and approved the supplier’s qualification protocol and reports for installation qualification (IQ) and operational qualification (OQ). All goes smoothly and you are ready to evaluate your new toy, when you hear the thud of heavy footsteps behind you. The Compliance Police have arrived to ruin your day!
The Compliance Police Cometh
The Compliance Police (Quality Assurance [QA] ± CSV) have descended and you are about to undergo the third degree questioning:
- Where is the system risk assessment?
- Where is the instrument user requirement specification?
- Have you approved the qualification against the user requirements?
- How did you evaluate the supplier quality system/approach to change control?
- Where is the software user requirements specification?
- Where is the requirements risk assessment?
- What is the software configuration?
- What user roles have been defined and implemented?
- Has QA signed off the supplier qualification documents before execution?
- Have you validated the software?
- Where is the validation summary report?
You can’t use the system until all these documents have been generated and approved. What?! All this documentation for a simple spectrometer that is just going to read absorption at specified wavelengths or verify the identity of a substance by comparison with a reference standard? Consider yourself lucky. You could have been asked to write a functional specification as well!
A risk-averse CSV approach can result in writing all these documents, especially if there is a one-size-fits-all procedure that is applied regardless of the intended use of the instrument and software. This is what gets CSV a bad name–inflexible organizations who just want to play it safe.
Integrated AIQ and CSV is Essential
Does it have to be this way? No.
The needs associated with the software and instrument hardware need to be addressed, and for simple intended use, this can be achieved in an integrated validation document.
To understand the approach, let’s start with regulations. We need to apply a 21 CFR 211.160(b) scientifically sound (1), flexible, common sense and logical approach to CSV. The key to doing this is understanding the 5 P’s:
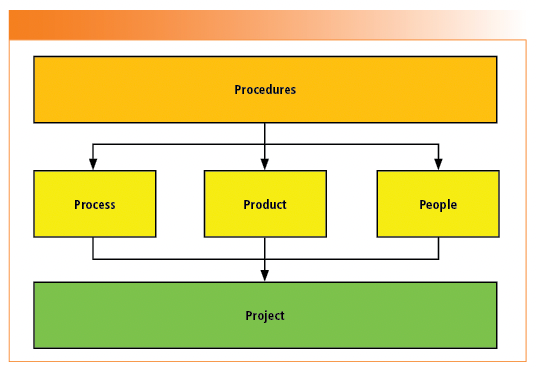
Procedures: Inputs are the regulations, regulatory guidance, and applicable pharmacopoeial general chapters that result in SOPs and analytical procedures for performing work. Flexible procedures that allow an integrated approach to Analytical Instrument Qualification (AIQ) and Computerized System Validation (CSV) are mandatory. Defining the intended use of the instrument or system (1,2) is the key to determining the required documents for qualification, validation, or both.
Process: Know and understand the process to be automated. Streamline and simplify the process, implement electronic signatures and eliminate dross such as paper and spreadsheets (3–5). This allows the laboratory to define the user requirements and configuration settings to ensure data integrity. Unfortunately, process redesign is not mentioned in the draft FDA Computer Software Assurance (CSA) guidance (6).
Product: The system consists of the instrument and the software that controls it. We must select the right system to digitalize a process with appropriate technical controls for data integrity. There is always an interaction between the product and the process to fine-tune configuration settings. Purchasing the right system that can digitalize a process and ensure data integrity must take precedence over purchasing on price. When application software is involved, it is critical that the supplier’s software development is assessed to see if it can be leveraged into the laboratory validation to reduce the effort required.
People: A multi-disciplinary team approach is required between key users, laboratory managers (Data Owner/Process Owner), QA, CSV and IT (System Owner), in addition to the supplier. The team must be trained, flexible, and manage risk effectively to qualify the instrument and validate the system, especially with the amount and extent of documentation required. When it comes to people, the role of the senior management is critical to lead and support the entire 5 P’s approach.
Project: The 4 P’s above come together in a project covering the definition, selection, qualification, and validation of the system. The scope and extent of the project depends on the intended use, process to be automated, records to be created and managed by the system.
The way the 5 P’s come together for an integrated approach is shown in Figure 1.


FIGURE 1: The 5 P’s of an integrated AIQ and CSV approach.
System Risk Assessment
To determine how much qualification and validation work needs to be performed, we need to time travel back to 2013, when Chris Burgess and I published a system risk assessment for analytical instruments and systems (7). One of the outcomes from the risk assessment was a sentence that referenced an article that described a single Integrated Validation Document (IVD) (8).
In essence, if the controlling software application is GAMP software category 3 and a commercially available non-configured product (9), even if it generates data used for analyses of clinical trial materials, product submissions or product releases, it could be validated using the IVD approach. Before discussing the IVD though, it is important to understand the difference between parameterization and configuration of software.
Parameterization Versus Configuration
Knowing how to differentiate between GAMP software categories 3 and 4 typically involves an arm wrestling match (sorry, collaboration) between the laboratory manager and CSV. Category 3 software cannot change the business process it automates; it does exactly what it says in the brochure, nothing more, nothing less. Although run time configuration is possible (for example, definition of user roles and access privileges, report formats), the business process cannot be changed.
A major cause of confusion is parameterization, which can be performed on functions in both category 3 and 4 software. Parameterization is setting the wavelength used to measure absorption of an analyte. If the wavelength is set at 220 nm and then changed to 280 nm, the business process has not changed. The instrument is still measuring the absorbance of an analyte regardless of wavelength, meaning the business process is unchanged. However, many people think this is configuration despite it being parameterization, so it still needs to be controlled when people use the system for things like method validation.
Principle for the Integrated Validation Document
EU GMP Annex 15 in clause 2.5 states:
Qualification documents may be combined together, where appropriate, e.g. installation qualification (IQ) and operational qualification (OQ) (10).
The principle of the IVD condenses the whole suite of validation documents into one. From the system risk assessment (7), this approach is only applicable to lower risk systems typically, but not exclusively, GAMP Category 3 software.
A description of the rational and justification can be found in the article by McDowall (8) which is available for free download (https://onlinelibrary.wiley.com/doi/10.1002/qaj.443).
Contents of an Integrated Validation Document
As the name suggests, all key validation requirements for demonstrating intended use are contained in a single document, as shown in Figure 2. An IVD has two main sections: specifications (shown in green) and testing/reporting (shown in yellow).

.")
FIGURE 2: Content of an integrated validation document (IVD).
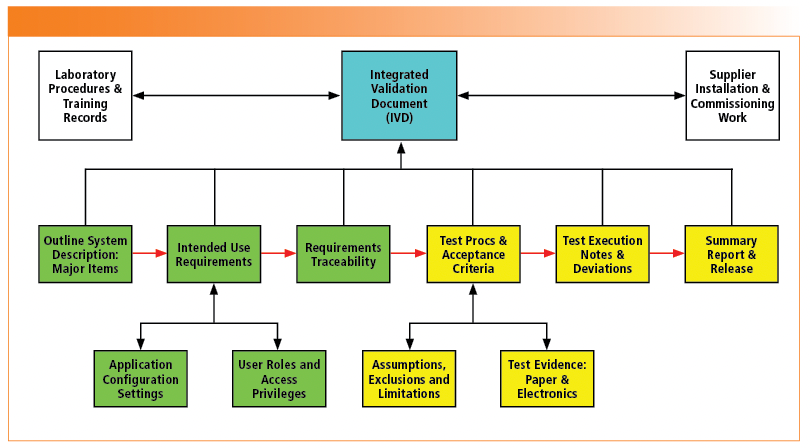
The size of a typical IVD is in the range of 30–45 pages, depending on the intended use of the system.
User Requirements and Configuration Specifications
The introduction and specifications, shown in green in Figure 2, consist of:
- System Description: A simple description of the intended use of the system along with a list of the main components e.g. instrument and software, standalone or networked system.
- Intended Use Requirements: Consisting of a series of tables consisting of three columns: requirement number, the requirement and a trace to where the requirement is met; e.g. test procedure, SOP, calibration, supplier documentation, and so forth. There are requirements tables for the function of the system, data integrity, IT support including protection of records, time synchronization, and more. If a user requirement is excluded from testing, this is justified in the section on assumptions, exclusions, and limitations.
- Specification of the system is limited to requirements only essential to achieve the intended use and are statements of what the system must do rather than what it can do. Therefore, just in case requirements are not considered, the discipline for an IVD is to focus purely on what the system will perform now rather than in the future.
- Raise a change request if additional requirements are needed when operational.
- Configuration Settings: A list or table of the application configuration settings for ensuring data integrity and non-functional settings. For example, unique user names, password expiry and complexity, and so forth.
- Definition of User Roles: Typically, this is a table of the various user roles against the access privileges allotted to each one.
Not shown in Figure 2 is a referenced documents section linking an IVD with regulations, CSV procedures, and applicable SOPs for operation of the system, such as user account management. In addition, supplier assessment and qualification documentation (for example, IQ, OQ, or calibration) will also be referenced and, where possible, used to verify user requirements.
Testing and Reporting of the Configured System
The second section of an IVD, colored yellow in Figure 2, consists of test procedures to confirm intended use, test execution notes to capture unexpected events during testing, and finishing with a reporting section with a simple release statement for operational use:
- Confirmation of Application Configuration: Confirmation that the configuration settings and policies of the application and user roles with access privileges correspond to those defined in the specification section of the IVD.
- Security testing of the system against user requirements.
- Intended Use Test Procedures: Requirements will define the tests to be performed to demonstrate the system’s intended use, the documented evidence anticipated (both electronic and paper), and acceptance criteria. The latter two are poorly defined in most validation examples I have audited.
- Test instructions are written for trained users to execute not idiots that require mind-blowing detail; see an earlier column for details (11). This enables sections to be written and executed quickly.
- If calculations are performed by the system, these must be verified as required by 21 CFR 211.68(b) (1) and Chapter 6.16 (12). One way will use a validated spreadsheet, but don’t forget to allow for differences in the number of significant figures when setting acceptance criteria.
- For the IT spectrometer used for identity testing, confirm that the compare function of the application works correctly.
- The system audit trail is explicitly tested using entries generated in earlier test procedures.
- IT support, such as backup and recovery, are also included in these test procedures.
- Test Preparation: Always remember you are testing software, not chemistry. Select analytical procedures that represent the intended use of the system and only use certified reference standards.
- Assumptions, Exclusions, and Limitations: As test procedures are developed by the team, it will become apparent why a test is being developed in a particular way. It is important to document any assumptions and limitations of the testing as well as why some requirements are excluded from testing, such as why expiry of passwords is not tested (13).
- Test Evidence Collation: The focus is on electronic records with as few as possible paper printouts as evidence of testing. There is a section in each test procedure for a tester to document this, making the review of the testing quicker and simpler to perform. Screenshots will be kept to an absolute minimum, as noted by the draft CSA guidance (6) and GAMP 5 2nd edition (9), and the focus is on the software, including the audit trail, to record evidence of testing. Though paper is a possible output, consider printing to PDF, as it can be a baseline for future data integrity audits and change control requests, such as configuration settings.
- Acceptance Criteria: Each test procedure has explicitly stated acceptance criteria that determine if testing has passed or failed.
- Test Execution Notes and Deviation Handling: If there are any problems or deviations when executing the test procedures, there is space in the IVD for documenting and resolving them.
The whole document is reviewed for technical content by the laboratory and approved by QA prior to execution.
Reporting and Release
After execution by the tester, they complete the Test Summary and Release Statement section of the IVD, stating whether or not the system can be released for operational use. The whole document is reviewed by a second person, who also confirms the release of the system. QA also reviews both the completed IVD and documented evidence.
Summary
You may think that this is a new approach to integrated qualification and validation of laboratory systems; however, it has been used for nearly 20 years. There have been minor changes in approach over the intervening years, but it is essentially the same as published. Go on, be lazy. You know it’s common sense, but will the Compliance Police let you?
Acknowledgments
I would like to thank Paul Smith and Mahboubeh Lotfinia for their constructive review comments in preparation of this article.
References
(1) Food and Drug Administration 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceutical Products. Silver Spring, MD, 2008.
(2) United States Pharmacopoeia Convention Inc. USP General Chapter <1058> Analytical Instrument Qualification. Rockville, MD.
(3) McDowall, R. D. Pharma 4.0 and the Digital Regulated Laboratory Part 1: Why Digitalize Your Regulated Laboratory? LCGC 2022.
(4) McDowall, R. D. Pharma 4.0 and the Digital Regulated Laboratory Part 2: Digital Laboratory Automation Strategy and Process Mapping. LCGC 2022.
(5) McDowall, R. D. Pharma 4.0 and the Digital Lab eBook 3: Quick Wins and Project Implementation. LCGC 2022.
(6) Food and Drug Administration. FDA Draft Guidance for Industry Computer Software Assurance for Production and Quality System Software. Silver Spring, MD, 2022.
(7) Burgess, C.; McDowall, R. D. An Integrated Risk Assessment for Analytical Instruments and Computerized Laboratory Systems. Spectroscopy 2013, 28 (11), 21–26.
(8) McDowall, R. D. Validation of Computerized Systems Using a Single Life Cycle Document (Integrated Validation Document). Qual. Assur. J. 2009, 12, 64–78. DOI: 10.1002/qaj.443
(9) International Society of Pharmaceutical Engineering. Good Automated Manufacturing Practice (GAMP) Guide 5, Second Edition. Tampa, FL, 2022.
(10) European Commission. EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Annex 15 Qualification and Validation. Brussels, Belgium, 2015.
(11) McDowall, R. D. Does CSA Mean “Complete Stupidity Assured?” Spectroscopy 2021, 36 (9), 15–22.
(12) European Commission. EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 6 Quality Control. Brussels, Belgium, 2014.
(13) McDowall, R. D. CSA: Much Ado About Nothing? Spectroscopy 2023, 38 (4), 7–13, 34.


R.D. McDowall is the director of R.D. McDowall Limited and the editor of the “Questions of Quality” column for LCGC Europe, Spectroscopy’s sister magazine. Direct correspondence to: SpectroscopyEdit@MMHGroup.com●











Synthesizing Synthetic Oligonucleotides: An Interview with the CEO of Oligo Factory
February 6th 2024LCGC and Spectroscopy Editor Patrick Lavery spoke with Oligo Factory CEO Chris Boggess about the company’s recently attained compliance with Good Manufacturing Practice (GMP) International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Expert Working Group (Q7) guidance and its distinction from Research Use Only (RUO) and International Organization for Standardization (ISO) 13485 designations.



