The Enhancement Effects of Forsythoside E on Serum Albumin Fluorescence Under Simulated Physiological Conditions
This study examines the fluorescence enhancement effects of forsythoside E, one of metabolites of Forsythia suspensa, on human serum albumin (HSA) and bovine serum albumin (BSA) under simulated physiological conditions. The increase in protein fluorescence induced by forsythoside E are approximately the same in slightly acidic and neutral solutions, whereas the protein fluorescence intensities barely change in the alkaline solution after adding forsythoside E. The fluorescence increase of proteins caused by forsythoside E are significantly bigger in ethanol than in the other two solvents. The enhancement effects of forsythoside E on protein fluorescence were weakened when Cu2+ and Fe3+ were pre-added to the solution. Forsythoside E had little effect on the fluorescence of glycosylated BSA (gBSA) but increased the fluorescence of glycosylated HSA (gHSA) in a concentration-dependent manner because BSA was significantly more glycosylated than HSA. These findings can help us to understand the delivering efficiency, absorption, toxicity, and the metabolic process of forsythoside E in vivo.
Serum proteins, the most abundant carrier proteins in plasma, are synthesized in the liver and can play a major role in delivering many endogenous and exogenous ligands by binding with them (1,2).
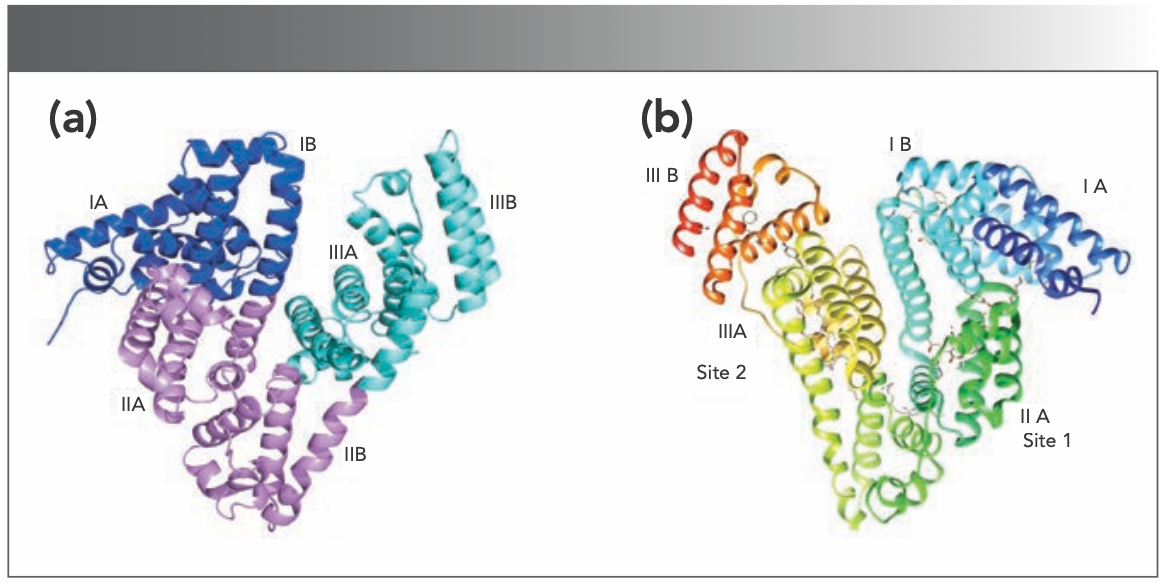
Human serum albumin (HSA) consists of 585 amino acids with a molecular weight of 66 kDa and contains only one tryptophan (Trp) residue (in this case, Trp 213). HSA, a monomeric globulin, has a heart-shaped stable structure of 17 pairs of disulfide bonds and one free cysteine residue. There are three domains (I, II, III) in HSA that encompass amino acid residues at positions 1–195, 196–383, and 384–585, respectively. Each domain includes two subunits (named A and B) that are connected by a long extended loop. The tertiary structure of HSA is composed of α-helix and a loop, forming pockets of binding sites at domains I–II and II–III to allow various endogenous and exogenous substances to bind strongly (3–6). The calculated net charges for HSA at a pH of 7 are −9, −8, and +2 in domains I, II, and III, respectively (7).
As a homologous protein with HSA, bovine serum albumin (BSA) has a 76% sequence resemblance with HSA and is widely used to study protein–drug interactions because of its well-known protein structure. BSA consists of 583 amino acid residues, including 20 tyrosine (Tyr) residues and two Trp residues (Trp 134 and Trp 213) that are responsible for the intrinsic fluorescence of BSA. Three specific homologous domains (I–III) form its main structure, known as 1–195, 196–383, and 384–583 amino acid residues, each of which is further divided into A and B subdomains. The BSA binding sites for ligands may be located in hydrophobic cavities in subdomains IIA and IIIA, known as Sudlow’s sites I and II, respectively. Trp 134 is situated on the surface of the protein, and Trp 213 is located within a hydrophobic binding pocket of the protein (8–11). The structures of HSA (6) and BSA (2) are shown in Figure S1 (Figures S1–S8 and Tables SI and SII can be accessed through the QR code found at the end of this article).

 bovine serum albumin (BSA) and (b) human serum albumin (HSA).")
FIGURE S1: Structures of (a) bovine serum albumin (BSA) and (b) human serum albumin (HSA).
The binding ability of serum proteins to drugs directly affects the delivery efficiency, distribution, absorption, excretion, stability, toxicity, and metabolism of drugs (1). Studying their mechanisms of action and interactions is important in understanding the pharmacokinetics of drugs. However, the presence of coexisting endogenous or exogenous substances and complicated internal environments often affect the ability of drugs to bind to proteins, which affects the free concentration and efficacy of drugs. Metal ions bind directly to serum proteins to weaken or enhance the binding of drugs to proteins. Cu2+ or Fe3+ reduced the quenching efficacy of rutin on BSA, increased the binding constant, and changed the binding distance between rutin and BSA that resulted from the formation of metal ion-BSA complex in regard to a combination of static quenching and nonradiative energy transfer (12). Thus, the increasing binding constant of rutin-BSA in the presence of the above ions prolonged the storage period of rutin in blood plasma and weakened its maximum effects (13). Glycation is a spontaneous nonenzymatic aminocarbonyl reaction between reducing sugars and long-lived proteins. Glycation is a major chemical modification in biomolecules and impairs the structural and functional characteristics of native serum proteins (14,15). Thus, the transportation, distribution, and function of drugs are affected strongly by the glycation of serum proteins (16). Differences in the pH environment have proven to have a great influence on the interaction between BSA and the compound (17).
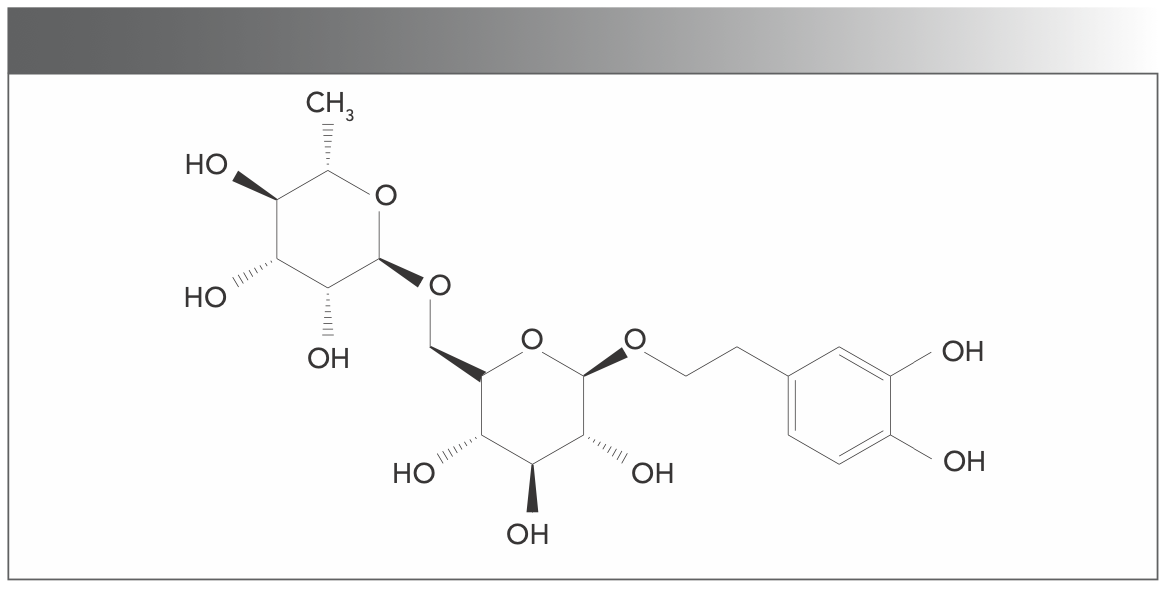
Forsythoside E (18) is one of two metabolites of F. suspensa that has been used as a traditional Chinese medicine to treat inflammation, based on its antioxidant, antibacterial, and antiviral activities (19). The structure of forsythoside E is shown in Figure S2. The interactions of forsythoside E and the two isoforms (forsythoside A and forsythoside I) from F. suspensa with BSA have been investigated in our group (20). Forsythoside E displayed results completely different from those of the other two isoforms; forsythoside E enhanced BSA fluorescence but the two isoformers quenched it. These novel results led us to further study the interactions of forsythoside E with serum albumin. Drugs intended for human use are first studied in simulated human body physiological conditions. Because proteins are denaturalized under specific conditions, they lose their original solubility and their physiological function is weakened. Proteins may behave differently in the body than they do outside the body, and the effect of drugs may be different. This study investigated and compared the interactions between forsythoside E, BSA, and HSA under minimal physiological conditions by using spectroscopic methods.


FIGURE S2: The structure of forsythoside E.
Material and Methods
Chemicals and Solution Preparation
Forsythoside E (with a purity of 98.6%) was purchased from Must Bio-Technology and was dissolved in purified water to prepare the stock solution. The final concentration of forsythoside E in the spectroscopic measurements was kept at 30 μM unless otherwise specified. BSA, HSA, CuCl2, FeCl3, and glucose were purchased from Solarbio and were made into a stock solution in purified water. The final concentrations of serum proteins in the spectroscopic measurements were kept at 1 μM unless otherwise specified. All other chemicals are analytical grade. All experiments were conducted in a Tris buffer with a pH of 7.4 except where designated.
Fluorescence Measurements
The fluorescence experiments were performed on a fluorescence spectrophotometer (Fluromax-4, Horiba Scientific). The temperature of the solution in the fluorescence cup was controlled at 27 ± 0.5 °C by a temperature holder, except where designated. The fluorescence between 290 and 500 nm was recorded at an excitation wavelength of 280 nm, with the fixed excitation and emission slit width of 10 nm each. Every time forsythoside E was added into the protein solution, the corresponding spectra were recorded 5 min later (the same protocol was carried out for all titration experiments).
To study the effects of Cu2+ and Fe3+ on the enhanced fluorescence of serum, proteins induced by forsythoside E, Cu2+, and Fe3+ were pre-added into the protein solution. After incubation, forsythoside E was gradually added to the mixture.
Synchronous Fluorescence Measurements
The synchronous fluorescence was recorded on the fluorescence spectrophotometer with the initial excitation wavelength of 220 nm and scanned up to 500 nm. The excitation and emission slit widths were fixed at 10 nm.
UV-vis Absorption Studies
UV-vis absorption spectra were recorded on a UV-vis spectrophotometer. The spectra were scanned between 200–350 nm. All the absorbance spectra were corrected by the blank.
Glycation of Serum Proteins
Glycosylated BSA (gBSA) and glycosylated HSA (gHSA) were prepared following a published method (14) with some changes. Serum proteins (the final concentration of 1 μM) were incubated with glucose (the final concentration of 1.1 mM) in a Tris buffer solution that had a pH of 7.4 and was kept at 37 °C for 10 days, with or without forsythoside E. After being dialyzed with the Tris buffer to remove the unbound glucose, the mixture was then used for spectrum measurement.
Circular Dichroism Measurements
Circular dichroism (CD) spectra were measured with a spectropolarimeter, which is equipped with a Quantum Northwest temperature controller.
Data Analysis
All data were expressed as means ± standard deviation. Each experiment was performed three times.
Results and Discussion
The Intrinsic Fluorescence of BSA and HSA in Different Environments
With the excitation wavelength of 280 nm, BSA and HSA have maximum emission peaks at approximately 348 nm, which is the characteristic emission wavelength of the Trp residue (21). Here, the intrinsic fluorescence of BSA and HSA in different environments was studied, and the fluorescence spectra are shown in Figure 1. The pH values vary greatly in the different organs and tissues in living systems, so the intrinsic fluorescence of BSA and HSA under different pH environments were investigated. The final concentrations of serum proteins were kept at 1 μM. Figure 1a shows that the fluorescence intensity of BSA changed little in a slightly acidic or neutral solution (pH 6.5, 7.0, and 7.4). Fluorescence intensities of BSA in an alkaline solution (pH 8.0, 8.5, and 9.0) were much lower than those in a slightly acidic and neutral solution and decreased gradually with the increase in alkalinity. HSA (Figure 1b) produced similar results. The position of the fluorescence emission peaks barely moved. Under the strongly basic conditions, proteins may denature and hydrolyze to amino acids.

 BSA and (b) HSA in different envi- ronments. The pH range was 6.5, 7.0, 7.4, 8.0 8.5 and 9.0. (c) Cu2+ at 0.5, 5, 10, and 30 μM and (d) Fe3+ at 0.5, 1, 5, and 10 μM decreased the intrinsic fluorescence of the proteins in a concentration dependent way; n = 3.")
FIGURE 1: The intrinsic fluorescence of (a) BSA and (b) HSA in different envi- ronments. The pH range was 6.5, 7.0, 7.4, 8.0 8.5 and 9.0. (c) Cu2+ at 0.5, 5, 10, and 30 μM and (d) Fe3+ at 0.5, 1, 5, and 10 μM decreased the intrinsic fluorescence of the proteins in a concentration dependent way; n = 3.
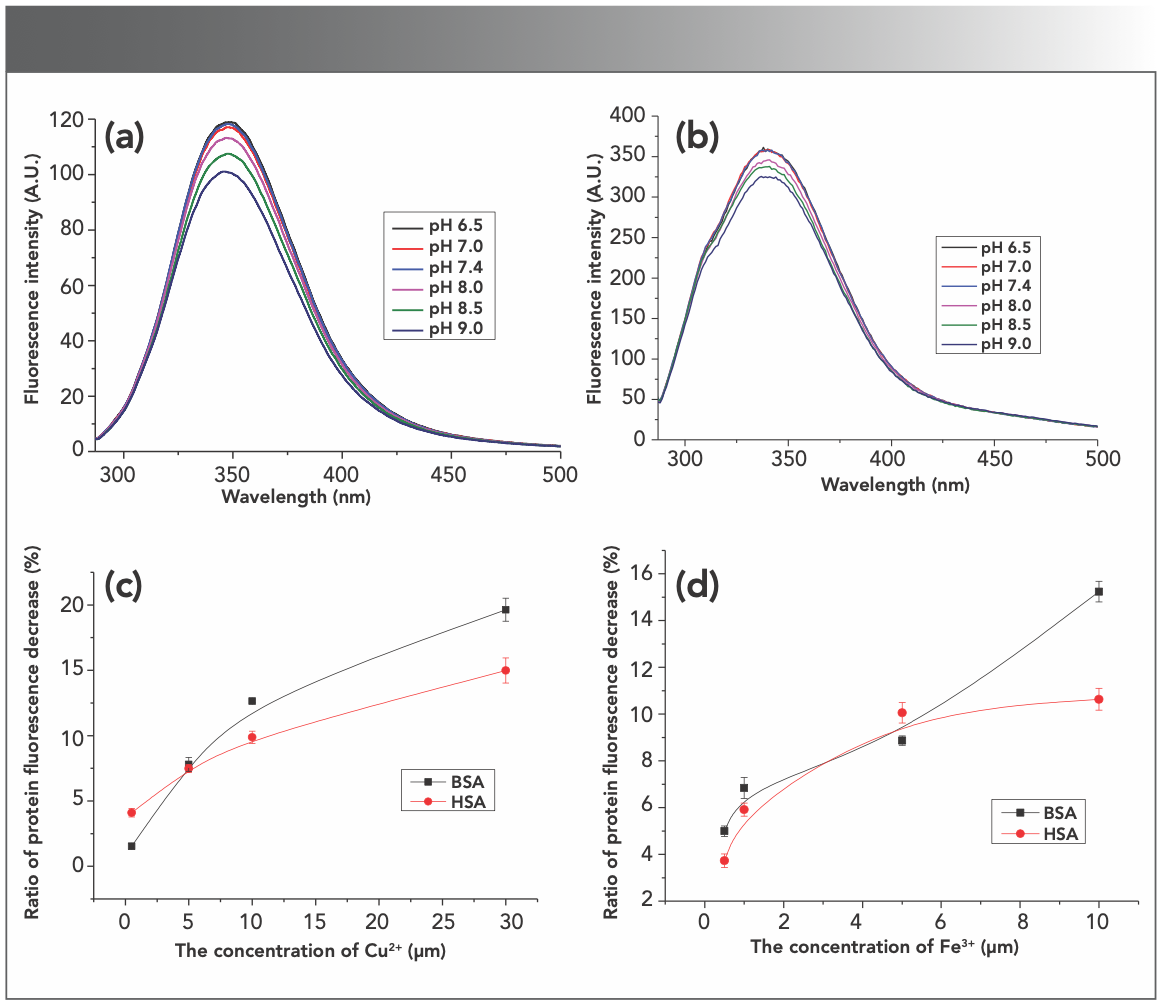
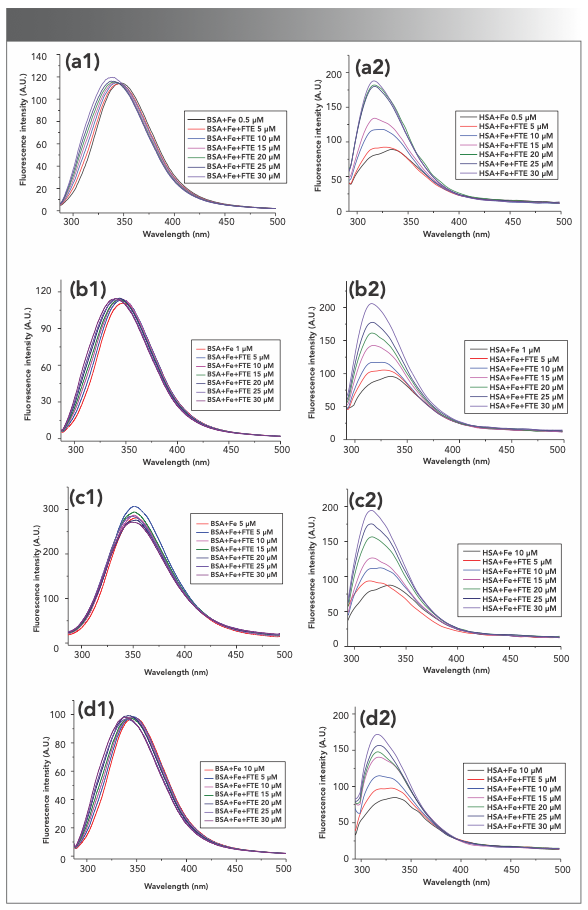
There are many metal ions in living systems that affect the structure and properties of proteins. Cu2+ and Fe3+ are the most common metal ions that are found in many organs and tissues. Cu2+, one of the unique and effective catalysts in biological systems, is the active component in more than 30 enzymes and plays an important role in metabolism of living systems (22). The effects of Cu2+ and Fe3+ on the intrinsic fluorescence of BSA and HSA were studied by adding metal ions into protein solution. As seen in Figure S3, Cu2+ (0.5–30 μM) and Fe3+ (0.5–10 μM) reduced the fluorescence intensities of BSA and HSA in a dose-dependent manner, and the reduction rate of BSA was higher than that of HSA (Figures 1c and 1d), which was consistent with the reported results (23). It suggested that the two metal ions may interact with BSA and HSA and the combination with the former was stronger than with the latter. Furthermore, the fluorescence of BSA may also be quenched by different concentrations of zinc (24), magnesium, or calcium ions (25).

 Cu2+ of 0.5–30 μM, and (b1 and b2) Fe3+ of 0.5–10 μM reduced the fluorescence intensities of BSA and HSA in a dose-dependent way.")
FIGURE S3: The effects of metal ions on the intrinsic fluorescence of BSA and HSA. (a1 and a2) Cu2+ of 0.5–30 μM, and (b1 and b2) Fe3+ of 0.5–10 μM reduced the fluorescence intensities of BSA and HSA in a dose-dependent way.
Fluorescence Enhancement of BSA and HSA Induced by Forsythoside E
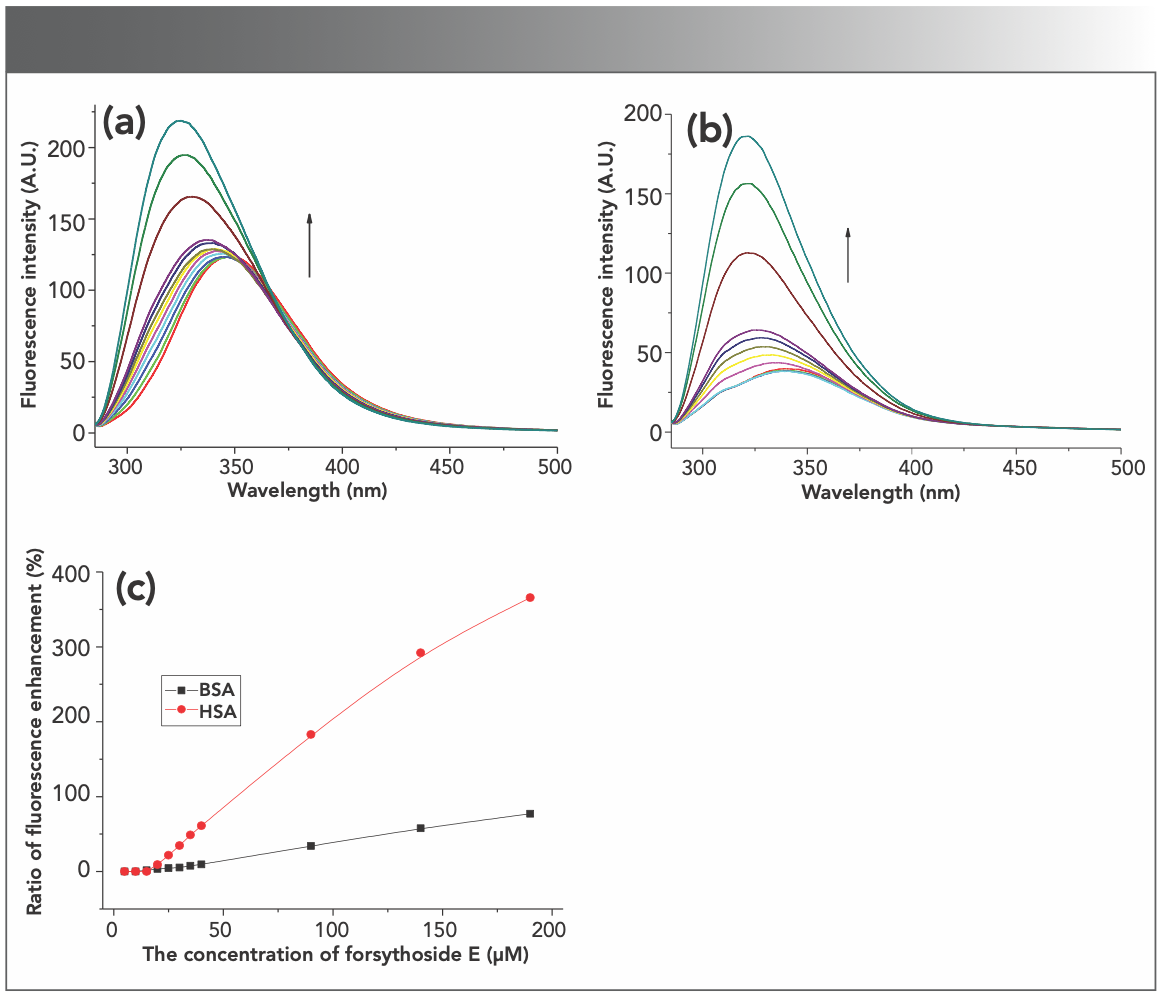
The change of intrinsic fluorescence of proteins induced by compounds is a signal to explore the interactions between compounds and proteins. The fluorescence of BSA and HSA was recorded after adding forsythoside E to serum protein solution. The final concentrations of serum proteins were kept at 1 μM in a Tris buffer with a pH of 7.4, and the final concentrations of forsythoside E were 5, 10, 15 . . . 40, 90, and 140 μM, respectively. The results were shown in Figures 2a and 2b. As the concentration of forsythoside E increased, the fluorescence intensities of BSA and HSA gradually increased, indicating that the compound interacted with the serum proteins. At the same time, the emission peak was blue-shifted, which suggested that the combination of the compound with the protein caused the protein peptide chain to fold further, strengthening its hydrophobic structure. As a result, the polarity around Trp decreased and hydrophobicity increased (24). The fluorescence enhancement rate of HSA induced by forsythoside E was much more than that of BSA (Figure 2c).

 BSA and (b) HSA fluorescence induced by forsythoside E. The concentrations of the proteins were kept at 1 μM. (c) The fluorescence enhancement rate of BSA and HSA induced by forsythoside E.")
FIGURE 2: The enhancement of (a) BSA and (b) HSA fluorescence induced by forsythoside E. The concentrations of the proteins were kept at 1 μM. (c) The fluorescence enhancement rate of BSA and HSA induced by forsythoside E.
The Enhancement Effects of Forsythoside E on BSA and HSA Fluorescence Under Different Conditions
The exogenous or endogenous nature of substances and different physiological environments in living systems may affect how drugs bind with serum albumin and are transported (12,13). The effects of forsythoside E on the intrinsic fluorescence of BSA and HSA were studied under the simulated physiological conditions. The data are listed in Table I.


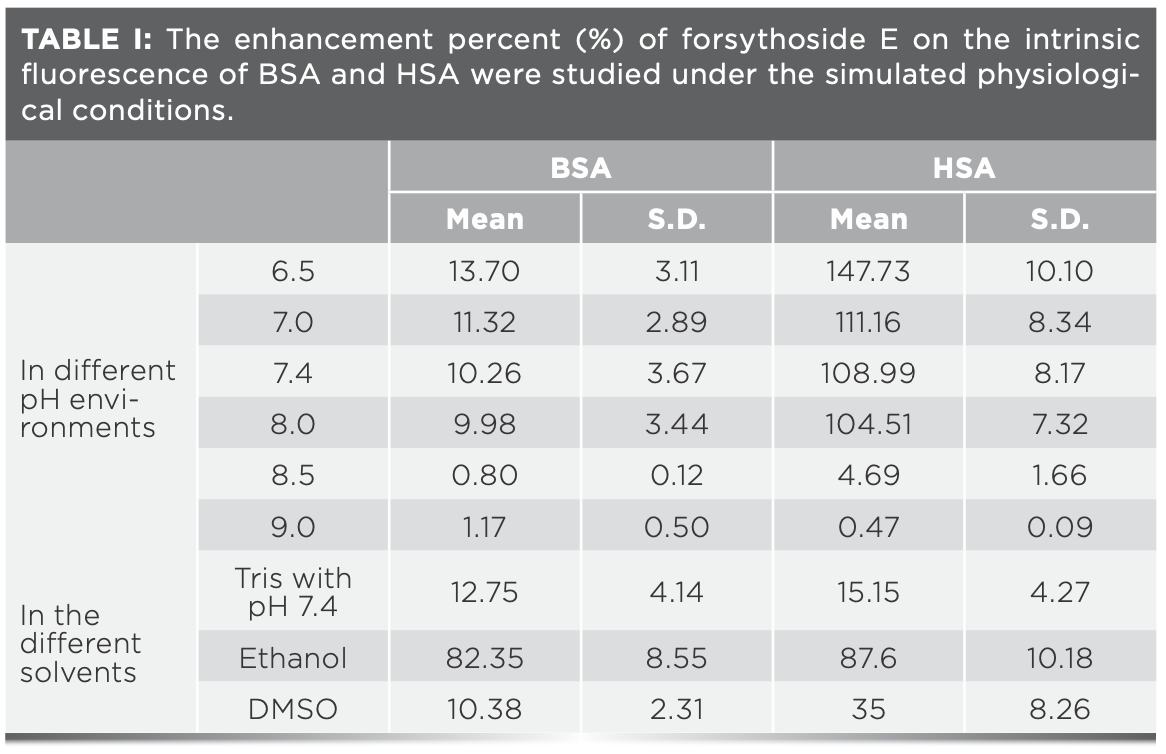
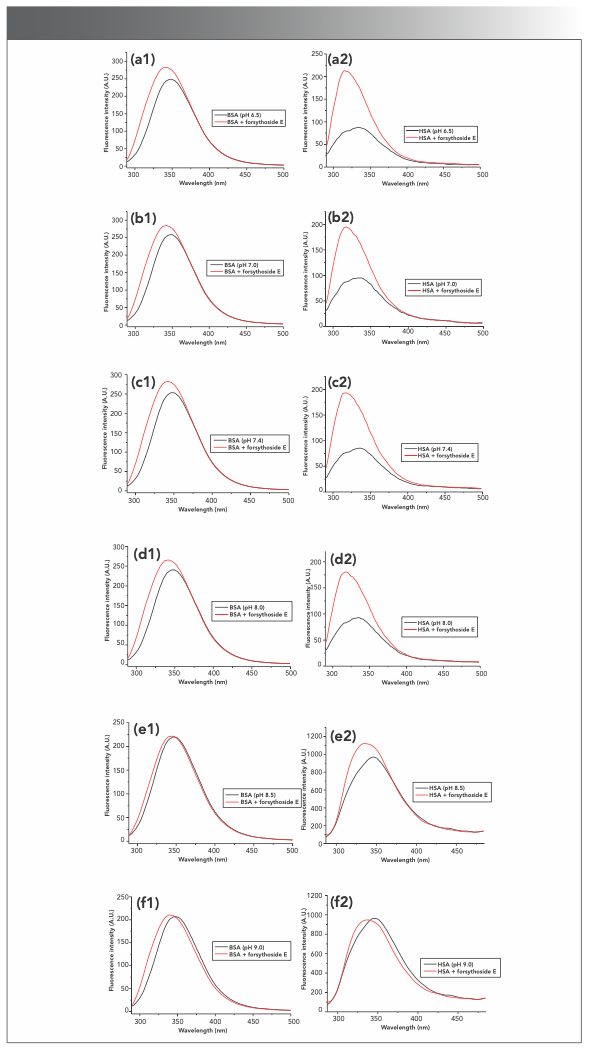
It was found that the increases in BSA fluorescence induced by forsythoside E were almost the same in slightly acidic and neutral solution (pH 6.5, 7.0, and 7.4). The fluorescence intensities of BSA in alkaline solution (pH 8.5 and 9.0) barely changed after adding forsythoside E to the solution. Similar results were found with HSA. However, the increases of HSA fluorescence that were induced by forsythoside E were much greater than the increases in BSA fluorescence under the same conditions. The fluorescence spectra are shown in Figure S4.

: BSA; (a2–f2): HSA.")
FIGURE S4: Effects of forsythoside E on the intrinsic fluorescence of BSA and HSA in different pH environments. The increases of protein fluorescence induced by forsythoside E were almost the same in slightly acidic and neutral solutions, whereas the protein fluorescence intensities in alkaline solution barely changed after adding forsythoside E. (a1–f1): BSA; (a2–f2): HSA.
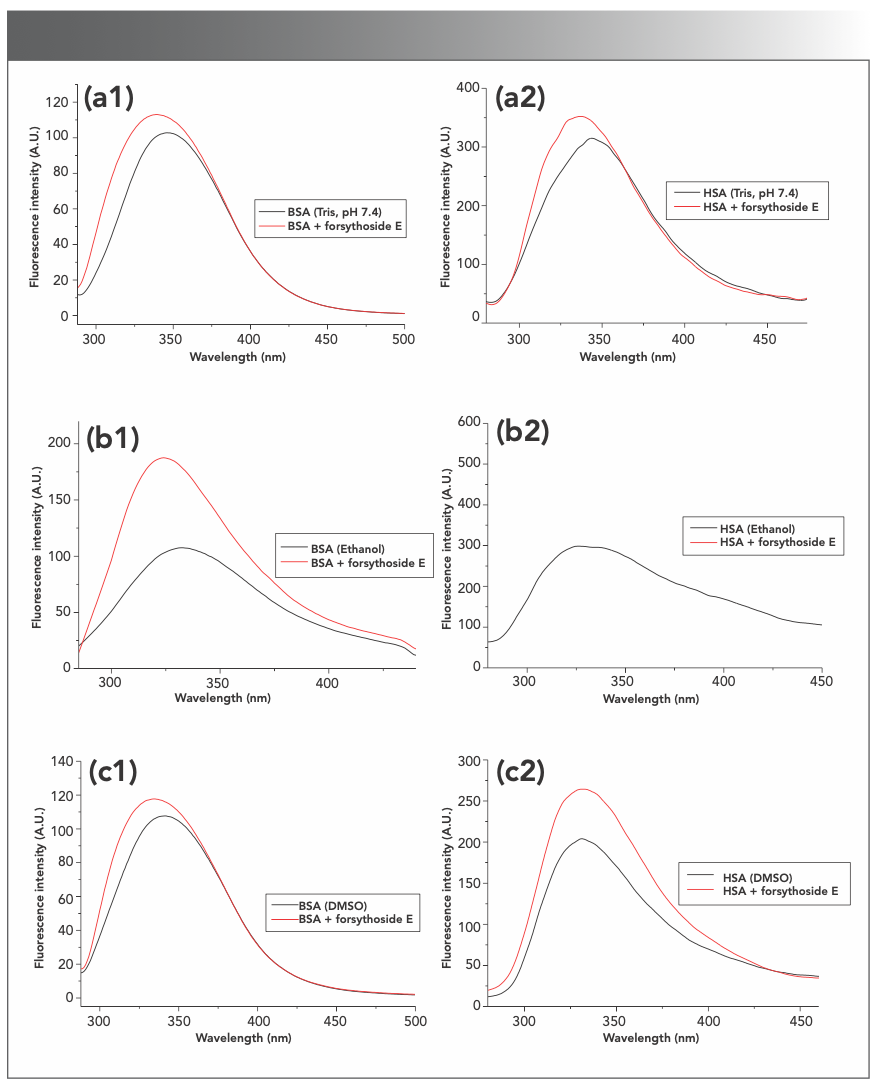
The change of protein fluorescence caused by forsythoside E in the different solvents was studied (Figure S5). The BSA fluorescence intensities increased with the addition of forsythoside E at the final concentration of 30 μM. The increase in BSA fluorescence induced by forsythoside E in the three solvents was the greatest in ethanol. HSA had similar results.

: BSA; (a2–c2): HSA.")
FIGURE S5: Effects of forsythoside E on the intrinsic fluorescence of BSA and HSA in the different solvents. The fluorescence increases of proteins that were caused by forsythoside E were much bigger in ethanol than those in the other two solvents. (a1–c1): BSA; (a2–c2): HSA.
Many metal ions in living systems affect the structures and properties of proteins. Cu2+ and Fe3+ are the most common metal ions in many organs and tissues. The effects of Cu2+ (Figure S6) and Fe3+ (Figure S7) on the fluorescence enhancement of BSA and HSA induced by forsythoside E were studied by adding metal ions to a protein solution. The results showed that the enhancement effect of forsythoside E on protein fluorescence was weakened after Cu2+ was pre-added to the protein solution. The intrinsic fluorescence of BSA barely changed at Cu2+ of 30 μM. We speculated several possible reasons. Serum protein has a limited number of binding sites. When Cu2+ was pre-added to BSA solution, Cu2+ was pre-added to BSA solution, Cu2+ combined with BSA and induced the conformational changes of BSA. Therefore, it was more difficult for forsythoside E to bind to BSA (12,13). The weakening effect of 2+ of 0.5–30 μM. With the increase of Fe3+ concentration, the enhancement effect of HSA fluorescence induced by forsythoside E gradually diminished and eventually disappeared. The study is helpful in understanding the effects of metal ions on the transportation, absorption, and release of forsythoside E in vivo.

: BSA; (a2–e2): HAS. Note: FTE is forsythoside E.")
FIGURE S6: Effects of forsythoside E on the intrinsic fluorescence of BSA and HSA in a Tris buffer with pH of 7.4 with the different concentrations of Cu2+. The enhancement effects of forsythoside E on protein fluorescence were weakened when Cu2+ was pre-added to the solution. a, b, c, d and e are 0, 0.5, 5, 10, and 30 μM of Cu2+, respectively. (a1–e1): BSA; (a2–e2): HAS. Note: FTE is forsythoside E.
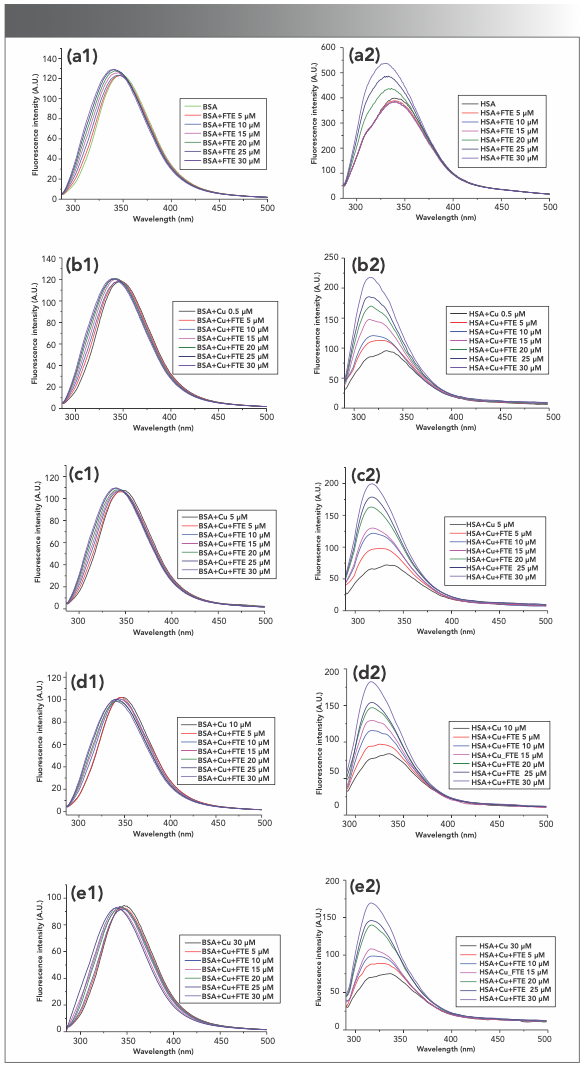

: BSA; (a2–d2): HSA. Note that FTE is forsythoside E.")
FIGURE S7: Effects of forsythoside E on the intrinsic fluorescence of BSA and HSA in a Tris buffer with a pH of 7.4 with the different concentrations of Fe3+. (a1–d1): BSA; (a2–d2): HSA. Note that FTE is forsythoside E.
Reducing sugar is another factor that affects the structures of serum proteins in the animal. Nonenzymatic glycation impairs the structural and functional characteristics of native serum proteins (14–16). The fluorescence of two serum proteins was recorded after glycation with glucose for 10 d and the results are shown in Figure 3a, where gBSA and gHSA represent glycosylated BSA and HSA, respectively. Compared to the fluorescence of the fresh native serum albumin, the fluorescence of gBSA markedly decreased, while the fluorescence of gHSA only slightly increased, suggesting that BSA was much more glycosylated than HSA. The effects of forsythoside E on the fluorescence of gBSA and gHSA were studied by adding forsythoside E into the glycosylated protein solution. Figure 3b shows that forsythoside E had little effect on the fluorescence of gBSA but increased the fluorescence of gHSA in a concentration-dependent manner.

 Fluorescence of BSA and HSA before and after glycosylation. (b) Effects of forsythoside E on glycosylated BSA (gBSA) and glycosylated HSA (gHSA). (c) The effect of forsythoside E on the secondary structures of proteins during glycosylation using CD spectra. (a1–c1): BSA. (a2–c2): HSA. Note FTE is forsythoside E.")
FIGURE 3: Effects of forsythoside E on glycation of BSA and HSA. (a) Fluorescence of BSA and HSA before and after glycosylation. (b) Effects of forsythoside E on glycosylated BSA (gBSA) and glycosylated HSA (gHSA). (c) The effect of forsythoside E on the secondary structures of proteins during glycosylation using CD spectra. (a1–c1): BSA. (a2–c2): HSA. Note FTE is forsythoside E.
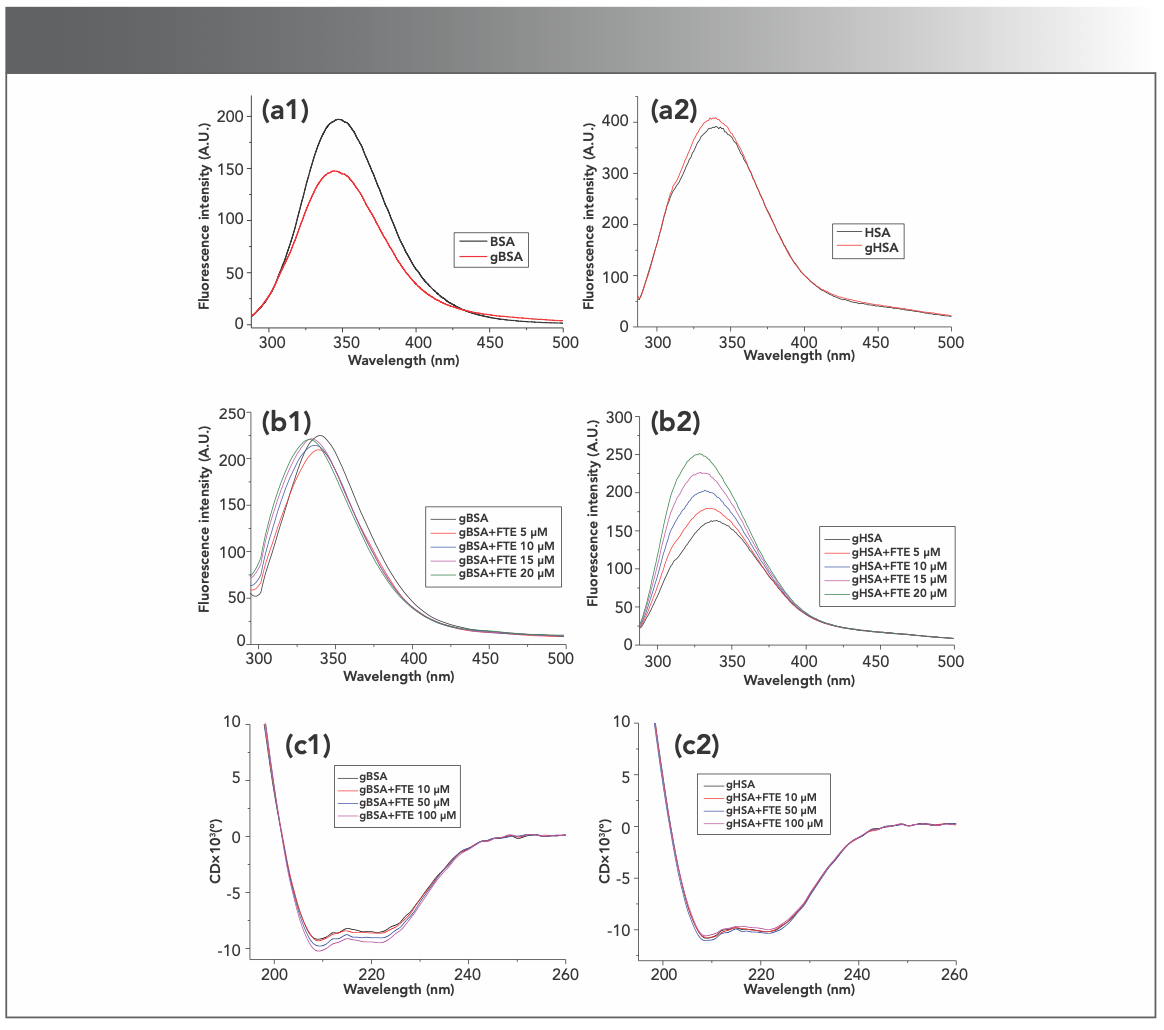
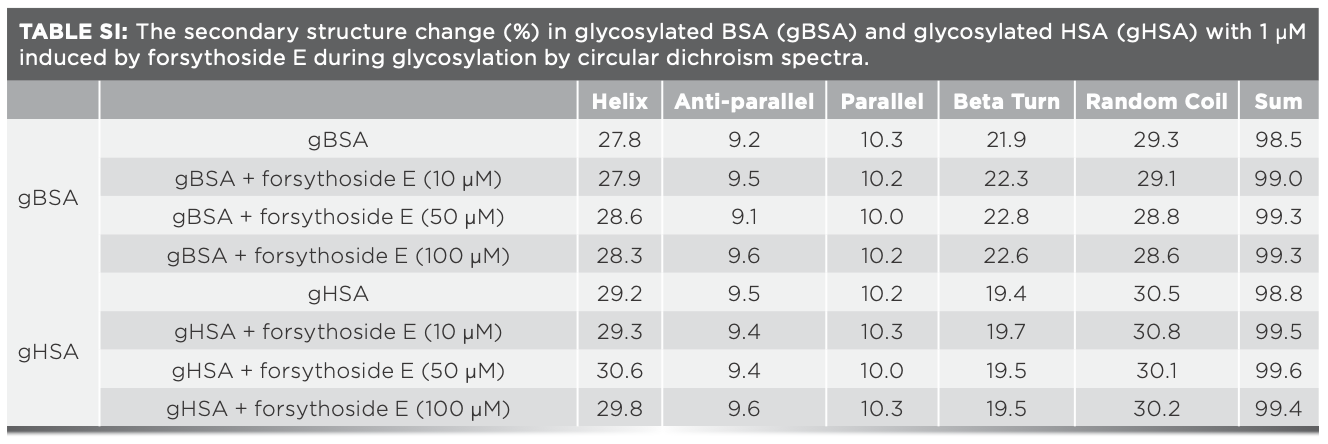
Next, the effects of forsythoside E on the secondary structure of gBSA and gHSA were studied by far-UV CD. Serum proteins (the final concentration of 1 μM) were incubated with glucose (the final concentration of 1.1 mM) in Tris buffer with pH 7.4 at 37 °C for 10 d, along with forsythoside E at the different concentrations. Far-UV CD spectra results are shown in Figure 3c. The negative peaks at 208 and 222 nm are two characteristic peaks of α-helix in serum proteins. Both peak values hardly varied for BSA and HSA with the forsythoside E at the different concentrations of 10 to 100 μM, which indicated no obvious change in the proportion of α-helix in the two serum proteins. The results suggest that forsythoside E inhibited the glycation of BSA and had no effect on the glycation of HSA (14–16,20). The percentage of α-helix, β-sheet, random coil, and other secondary structures of protein varied slightly (Table SI).


Fluorescence Enhancing Mechanism Studies
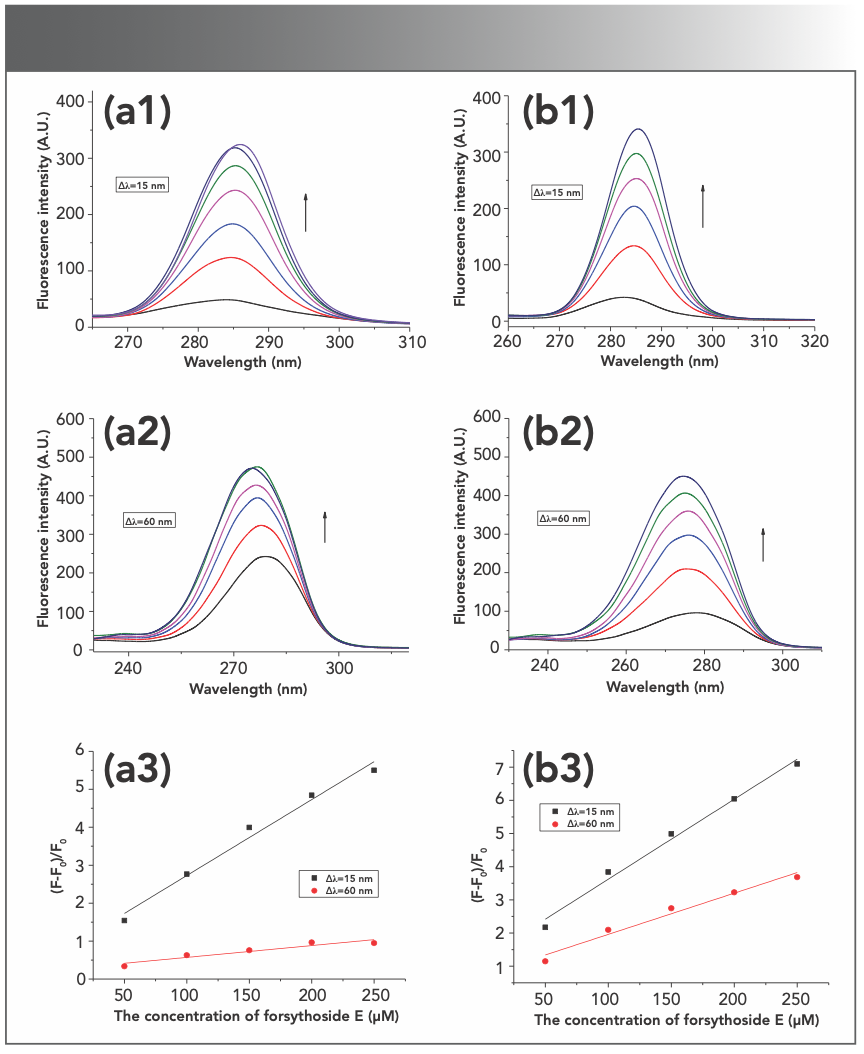
Synchronous fluorescence spectroscopy has been used to study changes in the microenvironment of amino acids in proteins. In that work, synchronous fluorescence was measured by adding forsythoside E to serum protein solution, with the concentrations of proteins of 1 μM and the concentrations of the compound of 50, 100, 150, 200, and 250 μM. As seen in Figure 4, the fluorescence intensities are regularly increased with the increase in the compound concentration, which further confirmed the fluorescence enhancement reported in the work. There was no significant shift in the maximum emission peak. The difference (λ) between excitation and emission wavelength at 15 nm and 60 nm represents Tyr residues and Trp residues in proteins, respectively (26). The fluorescence intensities of Tyr residues increase more, indicating that Tyr residues were more affected than Trp residues when the compound bound to serum proteins.

 BSA and (b1–b3) HSA.")
FIGURE 4: The synchronous fluorescence study of interaction of forsythoside E with (a1–a3) BSA and (b1–b3) HSA.
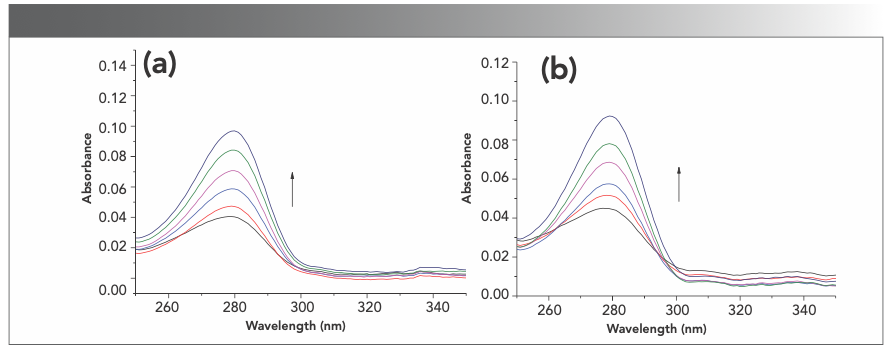
The protein–compound interaction was studied by addition of the compound (final concentrations of 5, 10, and 15 μM) to protein solution using a UV-vis absorption method. With the increase in compound concentration, the intensity of the absorption peak of proteins gradually increased, suggesting that a complex was formed between a protein and a compound by weak interaction (Figure S8).

 BSA and (b) HSA after the addition of forsythoside E at the different concentrations.")
FIGURE S8: The UV-vis absorption spectra of (a) BSA and (b) HSA after the addition of forsythoside E at the different concentrations.
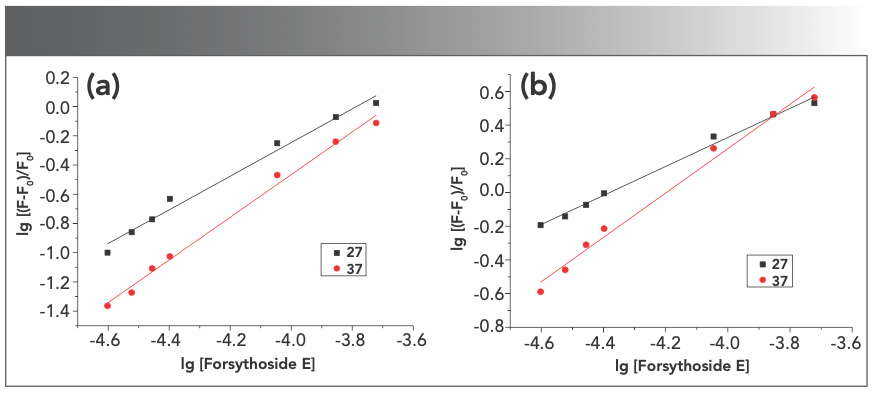
After knowing that the compound binds to BSA and HSA to form complexes, we calculated the values of the binding constant (K) and the number of binding sites (n) from the modified Stern-Volmer plots, namely, lg[(F-F0)/F0] vs. lg[Q] according to reported methods (27,28). The plots were shown in Figure 5. The K values of forsythoside E with BSA and HSA were both 1.45 × 104 1/M, with no significant differences, and the n values of forsythoside E with BSA and HSA were 1.09 and 0.95, respectively. Similar K and n values were reported for various albumin–drug systems (29,30). These results suggested that there was only one binding site in protein for the compound to combine to. The values of K and n for interactions between metal ions and proteins were much smaller than those between forsythoside E and proteins. However, the presence of metal ions still had significant effects on forsythoside E and protein interactions (31).

 BSA-forsythoside E and (b) HSA-forsythoside E.")
FIGURE 5: The logarithmic plots of (a) BSA-forsythoside E and (b) HSA-forsythoside E.
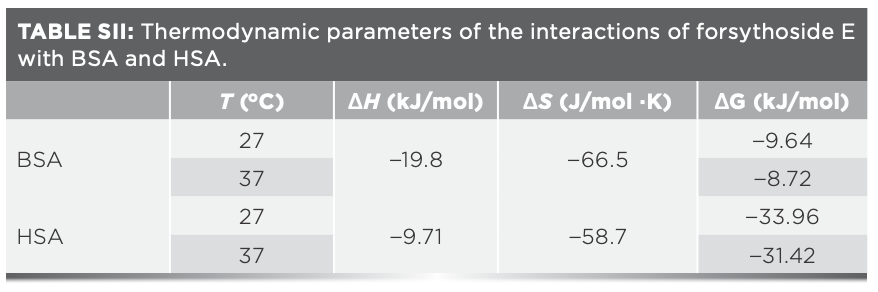
Four main types of weak and non-covalent interactions, including van der Waals forces, hydrogen bonds, electrostatic gravitational forces, and hydrophobic forces, play important roles in protein–compound interactions (32). The thermodynamic parameters ΔG, ΔH, and ΔS are always calculated by the equations and are then used to determine the type of binding forces involved in the protein–compound interaction process (21). In this work, the negative signs of all three parameters shown in Table SII reminded us that the interactions between forsythoside E and serum proteins occurred spontaneously and the main forces that formed forsythoside E–protein complexes were hydrogen bonds and van der Waals forces (33,34). The fluorescence spectra of the compound and the absorption spectra of the protein at the same concentrations did not precisely overlap in the same x-axis (figures not shown), so the binding distance (r) between them were not obtained. We speculate that nonradiative energy transfer was not the main cause of protein fluorescence enhancement induced by forsythoside E. These are consistent with other reports from our research group (20).


Conclusion
The domains of two serum proteins (BSA and HSA) exhibit extensive structural homology. However, their binding modes are not exactly the same. The interaction between a ligand and a protein may be competitive or cooperative. In this work, forsythoside E increased the intrinsic fluorescence of proteins in the form of a protein–forsythoside E complex with a stoichiometric ratio of 1:1. The increases in protein fluorescence induced by forsythoside E were almost the same in slightly acidic and neutral solutions, whereas the protein fluorescence intensities in alkaline solutions barely changed after adding forsythoside E. Among the three solvents, the increase of serum fluorescence caused by forsythoside E was the greatest in ethanol. The enhancement effect of forsythoside E on protein fluorescence was weakened when Cu2+ and Fe3+ were present in the protein solution. BSA was confirmed to be more glycosylated than HSA, so forsythoside E had little effect on the fluorescence of gBSA but increased the fluorescence of gHSA in a concentration-dependent manner. These findings will contribute to our understanding of the transportation, delivery efficiency, absorption, toxicity, and metabolic process of forsythoside E in vivo.
Conflict of Interest Statement
The authors declare that they have no conflict of interest.
Acknowledgments
This work was supported by Shanxi Scholarship Council of China (No. 2017-021), Natural Science Foundation of Shanxi Province (No. 201801D121290) and Students Research Training for undergraduates of Shanxi University (No. 2019017314). We thank Scientific Instrument Center of Shanxi University for the technical support.
References
(1) M.J. Banker and T.H. Clark, Curr. Drug Metab. 9, 854–859 (2008).
(2) A. Bujacz, Acta Crystallogr. D Biol. Crystallogr. 68, 1278–1289 (2012).
(3) X.M. He and D.C. Carter, Nature 358, 209–215 (1992).
(4) G. Rabbani and S.N. Ahn, Int. J. Biol. Macromol. 123, 979–990 (2019).
(5) Y. Ishima and T. Maruyama, Yakugaku Zasshi. 136, 39–47 (2016).
(6) J. Aslam, I.H. Lone, N.R.E. Radwan, M.F. Siddiqui, S. Parveen, R.B. Alnoman, and R. Aslam, ACS Omega 4, 22152–22160 (2019).
(7) S. Tunç, O. Duman, and B.K. Bozoğlan, J. Lumin. 140, 87–94 (2013).
(8) K.A. Majorek, P.J. Porebski, A. Dayal, M.D. Zimmerman, K. Jablonska, A.J. Stewart, M. Chruszcz, and W. Minor, Mol. Immunol. 52, 174–182 (2012).
(9) Y. Ni, R. Zhu, and S. Kokot, Analyst 136, 4794–4801 (2011).
(10) N. Tayeh, T. Rungassamy, and J.R. Albani, J. Pharm. Biomed. Anal. 50, 107–116 (2009).
(11) D.C. Carter and J.X. Ho, Adv. Protein Chem. 45, 153–203 (1994).Adv. Protein Chem.
(12) D. Li, M. Zhu, C. Xu, and B. Ji, Eur. J. Med. Chem. 46, 588–599 (2011).
(13) D. Li, M. Zhu, C. Xu, J. Chen, and B. Ji, Spectrochim. Acta A Mol. Biomol. Spectrosc. 78, 74–79 (2011).
(14) N. Sattarahmady, A.A. Moosavi-Movahedi, F. Ahmad, G.H. Hakimelahi, M. Habibi-Rezaei, A.A. Saboury, and N. Sheibani, Biochim. Biophys. Acta 1770, 933–942 (2007).
(15) A. Szkudlarek, M. Maciążek-Jurczyk, M. Chudzik, J. Równicka-Zubik, and A. Sułkowska, Spectrochim. Acta A Mol. Biomol. Spectrosc. 153, 560–565 (2016).
(16) A. Raghav, J. Ahmad, and K. Alam, Diabetes Metab. Syndr. 10, 96–101 (2016).
(17) A.M. Siddiq, D. Murugan, R. Srivastava, and Md S. Alam, Colloids Surf B Biointerfaces 184, 110524 (2019).
(18) K. Endo and H. Hikino, Can. J. Chem. 62, 2011–2014 (1984).
(19) Chinese Pharmacopoeia Commission, Pharmacopoeia of the People’s Republic of China, Part One (China Medical Science Press, Beijing, P.R. China, 2015), pp. 170–171.
(20) Y. Li, Q. Guo, Y. Yan, T. Chen, C. Du, and H. Du, Spectrochim. Acta A Mol. Biomol. Spectrosc. 214, 309–319 (2019).
(21) P. Alam, A.S. Abdelhameed, R.K. Rajpoot, and R.H. Khan, J. Photochem. Photobiol. B 157, 70–76 (2016).
(22) Y. Park, C.Y. Lee, K.S. Park, and H.G. Park, Chem. Eur. J. 23, 17379–17383 (2017).
(23) X. Xu, L. Zhang, D. Shen, H. Wu, and Q. Liu, J. Fluoresc. 18, 193–201 (2008).
(24) X.Y. Jiang, W.X. Li, and Q.J. Chen, Chin. J. Inorg. Chem. 2008, 1588–1595 (2008).
(25) A.J. Stewart, C.A. Blindauer, S. Berezenko, D. Sleep, and P.J. Sadler, Proc. Natl. Acad. Sci. U.S.A. 100, 3701–3706 (2003).
(26) J.N. Miller and A.F. Fell, J. Pharm. Pharmacol. 32, 70 (1980).
(27) G.A. Siddiqui, M.K. Siddiqi, R.H. Khan, and A. Naeem, Spectrochim. Acta A Mol. Biomol. Spectrosc. 203, 40–47 (2018).
(28) B. Wu, C. Qu, Y. Wang, J. Zhao, and H. Du, J. Fluoresc. 29, 1113–1123 (2019).
(29) O. Duman, S. Tunç, and B.K. Bozoğlan, J. Fluoresc. 23, 659–669 (2013).
(30) S. Tunç, A. Cetinkaya, and O. Duman, J. Photochem. Photobiol. B 120, 59–65 (2013).
(31) L. Liu, G. Fang, and G. Song, Anal. Instrum. 2005, 18–21 (2005).
(32) I.M. Klotz, Ann. N. Y. Sci. 226, 18–35 (1973).
(33) M. Ishtikhar, E. Ahmad, Z. Siddiqui, S. Ahmad, M.V. Khan, M. Zaman, M.K. Siddiqi, et al, Int. J. Biol. Macromol. 107, 2450–2464 (2018).
(34) X. Yan, T. Chen, L. Zhang, and H. Du, Int. J. Biol. Macromol. 119, 1344–1352 (2018).
Li Dang is with the Shanxi Pharmaceutical Vocational College, in Taiyuan, China. Xuemei Li, Sifan Guo, and Huizhi Du are with the Institute of Molecular Science at Shanxi University, in Taiyuan, China. Direct correspondence to: duhuizhi@sxu.edu.cn.●

 Prove? A Look at Inorganic Analyses, Proficiency Tests, and Contamination and Error, Part I")







Geographical Traceability of Millet by Mid-Infrared Spectroscopy and Feature Extraction
February 13th 2025The study developed an effective mid-infrared spectroscopic identification model, combining principal component analysis (PCA) and support vector machine (SVM), to accurately determine the geographical origin of five types of millet with a recognition accuracy of up to 99.2% for the training set and 98.3% for the prediction set.



Authenticity Identification of Panax notoginseng by Terahertz Spectroscopy Combined with LS-SVM
In this article, it is explored whether THz-TDS combined with LS-SVM can be used to effectively identify the authenticity of Panax notoginseng, a traditional Chinese medicine.

