Validating ICP-MS for the Analysis of Elemental Impurities According to Draft USP General Chapters <232> and <233>
The current United States Pharmacopeia (USP) General Chapter <231> "heavy metals limit test" will be replaced by new instrumental methods, USP <232> (Limits) and <233> (Procedures), in 2013.
The current United States Pharmacopeia (USP) General Chapter <231> "heavy metals limit test" will be replaced by new instrumental methods, USP <232> (Limits) and <233> (Procedures), in 2013.
The presence of impurities in pharmaceutical samples is a concern, not only because of potential toxicity, but also because of the potential impact on drug stability, shelf-life, or unwanted side-effects. As a result, both organic and inorganic (elemental) impurities must be monitored and controlled in raw materials used for drug manufacturing, in intermediates and active pharmaceutical ingredients (APIs), in excipients (stabilizers, fillers, binders, colorings, flavors, coatings, and so forth), and in final drug products. Impurities that may be added during the production process, such as catalysts and contaminants from production process equipment, must also be monitored.
The current method used for monitoring inorganic contaminants in pharmaceutical samples is a 100-year-old colorimetric test, defined in United States Pharmacopeia (USP) General Chapter <231>. This method, known as the "heavy metal limit test," is not specific for individual analytes, and does not include many elements of interest, such as Cr and the catalyst metals.
Another acknowledged problem with USP <231> is that the sample preparation method requires ignition of the sample at temperatures up to 600 °C, which leads to loss of volatile analytes, including the critical toxic element Hg (1–4).
USP is currently developing two new General Chapters USP <232> (Limits) and <233> (Procedures), which will be implemented in 2013. The new methods will address the limitations of the current method, especially the list of analytes and the use of closed-vessel sample digestion and modern instrumental techniques for the accurate recovery and determination of individual analyte concentrations.
Table sI (available online) shows the permitted daily exposure (PDE) limits for the new list of 16 analytes (As, Cd, Hg, Pb, V, Cr, Ni, Mo, Mn, Cu, Pt, Pd, Ru, Rh, Os, and Ir) defined in USP <232> (5). This analyte list was developed based on toxicological data, rather than method capability, and for the first time the list includes catalyst elements (the platinum group elements [PGEs] Pt, Pd, Ru, Rh, Os, and Ir). USP <232> also contains a section relating to the elemental form (species) of elements, and notes that As and Hg are of particular concern as some forms are much more toxic than others.



Table sI: USP permitted daily exposure (PDE) limits for drug products and concentration limits for components (drug substances and excipients) of drug products with a maximum daily dose â¤10 g/day (5), together with ICP-MS system detection limits.
Although the required PDE limits defined in USP <232> can be easily measured with either of the instrumental techniques referenced in USP <233> (inductively coupled plasma–mass spectrometry [ICP-MS] or inductively coupled plasma–optical emission spectrometry [ICP-OES]) (6), many novel drugs may only be available in very small amounts. The large dilution associated with the preparation of these mg-scale sample weights means that instrumentation with the lowest possible detection limits may be essential.
USP <232> also provides individual component limits for drug substances and excipients, assuming a maximum daily dose not more than 10 g/day. These component limits are applicable to manufacturing quality control as they allow drug manufacturers to control the concentration of impurities in the raw materials and intermediates used in the final drug product. For any sample requiring digestion or dilution, the PDE limits must be corrected for the dilution factor applied during sample preparation. For example, the individual component limit for Cd is 0.5 µg/g (ppm) for solid drug products and excipients. A dilution factor of 250× during sample digestion would give a PDE limit in the sample digest (the "J" value) of 2 ng/mL (ppb) for Cd. Accurate recovery must be demonstrated at 0.5J (1 ng/mL), suggesting a required detection limit at least 10× lower than this (0.1 ng/mL) a concentration easily measured using ICP-MS, as shown in Table sI.
The limits shown in Table sI are based on oral administration and also are modified depending on the route of administration. Component limits for drug products and excipients that would be delivered by parenteral or inhalational administration must be 10× lower than these values, suggesting a required detection limit of 0.01 ng/mL, still easily within the range of ICP-MS.
A wide range of possible samples may be analyzed using USP <232> and <233>. Some pharmaceutical samples can be analyzed directly (unsolvated), while others can be prepared using simple dilution or solubilization in an aqueous solvent (such as water or dilute acid) or a suitable organic solvent (such as 25:75 2-butoxyethanol–water [3], DMSO, or DGME). For many APIs, dilution in an organic solvent is the preferred approach, in which case it may be necessary to include some means of stabilizing the analytes to avoid variable recovery due to the presence of more or less volatile species compared to the calibration standard (7).
USP <233> defines the sample preparation and method validation procedures that should be used for system suitability qualifications to ensure that the analysis is "specific, accurate, and precise" (6).
In practice, most laboratories are likely to opt for one of the ICP techniques, due to their ability to handle a wide range of sample types and to measure all required elements in a single rapid analysis (8,9).
In this study, we performed method validation and system suitability performance testing of ICP-MS for the analysis of pharmaceutical products, according to the May 2011 revision of USP <232> and <233>.
Methods
Sample Preparation
Many raw materials, excipients, intermediates, APIs, and final products will be insoluble in aqueous or organic solvents, and will therefore require acid digestion. USP <233> specifies the use of "strong acids" for digestion and there are some general points that will apply to most sample types that require digestion:
- The list of elements in USP <232> includes Hg and the PGEs. These elements are chemically unstable in an oxidizing matrix, such as nitric acid (HNO3) or nitric/peroxide (HNO3/H2O2) (10,11), and can only be stabilized and measured reliably over an extended period if the digest solution includes a complexing agent such as HCl. We prepared all samples using a digestion matrix of 1% HNO3 and 0.5% HCl to ensure stability of Hg and the PGEs.
- Pharmaceutical products may be a complex combination of the API, plus fillers, binders, colorings, and coatings. These coatings may be organic polymers that are formulated to resist acid attack in the stomach and thereby control the point at which the drug substance is released in the small intestine. Closed-vessel (high temperature and pressure) microwave digestion is the preferred digestion technique referred to in USP <233> for solid samples. Closed-vessel digestion also eliminates any issues of loss of volatile elements such as Hg, which is a problem with USP <231>.
Instrumentation
All 16 elements specified in the May 2011 revision of USP <232> were measured using ICP-MS. The full suite of analytes plus internal standards was measured in calibration standards, multiple replicates of digested gelatin capsule (gelcap) samples, and samples spiked at the levels specified in USP <233>. The ICP-MS system provided a high plasma temperature (low CeO/Ce ratio of around 1%), which delivered good matrix decomposition and high sensitivity for poorly ionized elements such as As, Cd, Hg, and the PGEs: Os, Ir, and Pt. The system also includes a Peltier-cooled spray chamber, which is preferred in USP <233>.
The ICP-MS system was operated in helium (He) collision mode, which eliminates matrix-based polyatomic interferences regardless of sample composition and without the sample-specific or analyte-specific optimization that is a characteristic of reaction cell methods (12). He mode operation allows samples that contain high and variable amounts of chloride to be run without compromising the detection of elements that can suffer from chloride-based polyatomic overlaps. These elements include 51V (overlap from 35Cl16O), 52 Cr (35Cl16O1H),53 Cr (37Cl16O), and 75As (40Ar35Cl). The operating conditions used for the analysis of gelcap sample digests are shown in Table II.


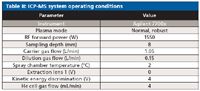
Table II: ICP-MS system operating conditions
An additional benefit of He mode operation is that it removes the polyatomic interferences from all isotopes of each analyte, so secondary or qualifier isotopes are available for analyte confirmation. USP <233> specifies that the procedure must be able to unequivocally assess each target element in the presence of other sample components such as other analytes and matrix components. The use of secondary isotopes as qualifier ions for ICP-MS is a well-established and unique capability of He mode (13).
Analytical Method
A standard 7700x ICP-MS system from Agilent Technologies (Santa Clara, California) was used throughout. The collision–reaction cell (CRC) ICP-MS uses an octopole reaction system (ORS3 ) for removal of polyatomic interferences. The ORS was operated in He mode for all analytes and all samples, demonstrating the simple method setup and routine operation provided by ICP-MS. Primary and secondary (qualifier) isotopes, cell mode, integration times, and method detection limits (MDLs) for all analytes are shown in Table sIII (available online). The MDLs were calculated from threefold the standard deviation of 10 external measurements of unspiked gelcap samples.


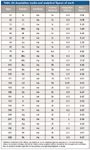
Table sIII: Acquisition modes and analytical figures of merit
External calibrations were prepared from single-element ICP-MS stock solutions at 1000 ppm. Because several of the target elements are not stable unless HCl is present in the solution, the intermediate stock solution was prepared in the same acid mix used for the final sample dilutions (1% HNO3 and 0.5% HCl). Example calibration plots of the four most critical elements As, Cd, Hg, and Pb are shown in Figure s1 (available online). Good linearity and sensitivity were demonstrated for all elements, and the low background equivalent concentration (BEC) for As demonstrated the effective removal of the ArCl polyatomic interference.



Figure s1: Calibrations for As, Cd, Hg, and Pb demonstrating excellent linearity and low backgrounds.
Results
System Suitability Check and Method Validation
The validation procedure described in USP <233> requires that a standardization solution at 2J (two times the control limit corrected for sample dilution) is measured before and after the sample batch. The drift between these two solutions must not exceed 20%.
To confirm the long-term stability of the results we used the 2J standard as a continuing calibration verification (CCV) quality control (QC) check run periodically during the 7 h sequence. The results for the sequence are summarized in Table sIV (available online), demonstrating that the initial calibration remained valid with overall drift from the first to last CCV of less than 7.5% for all elements and less than 2% for many. This demonstrates the robustness of ICP-MS for the routine analysis of pharmaceutical samples.


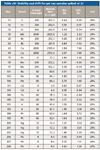
Table sIV: Stability and drift for gel cap samples spiked at 2J
The method validation requirements of USP <233> depend on the procedure used and whether it is a limit procedure or a quantitative procedure. Limit procedures must confirm detectability, repeatability, and specificity of the measurement, and quantitative procedures must demonstrate accuracy, precision (repeatability and ruggedness), and specificity.
Limit procedures must be capable of detecting a sample spike by comparing the measured result for a sample spiked at 1J, against a standard solution prepared at 1J. The spiked sample mean result must be the same as (within ±10%) or greater than the value obtained in the standard solution. The procedure must also be able to distinguish between a test sample spiked at the target concentration (1J) and a sample of the same material spiked at 80% of the target concentration (0.8J). The result for the sample spiked at 0.8J must be less than the result for the sample spiked at 1J. Repeatability must be demonstrated by meeting an external precision limit of no more than 20% RSD for six separate samples spiked at the spike levels.
Results for the limit procedure validation of gelcaps measured using ICP-MS are shown in Table V. The mean results for six external repeats of the standard at 1J, six gelcap samples spiked at 1J, and six gelcap samples spiked at 0.8J are presented. The results confirm excellent accuracy and repeatability, easily within the validation requirements for both spike recovery and discrimination. Specificity is also demonstrated through the use of secondary isotopes to confirm unequivocal assessment of each target element in the presence of other analytes and the matrix components.



Table V: Accuracy of spike recovery at 1J in gel caps, and discrimination between spikes at 1J and 0.8J (n = 6 for each sample)
Quant procedures must demonstrate accurate spike recovery (between 70% and 150% of the spike value) for the mean of three samples spiked at concentrations ranging from 50% to 150% of the J value (0.5J to 1.5J) for each target element. In this validation, we used spikes at 0.5J and 1.5J, as well as the 0.8J and 1J spikes measured for the limit procedure. Six separate (external) samples were analyzed for each spike level.
Repeatability must be demonstrated by an external precision not more than 20% RSD for six separate samples spiked at the indicated levels. Ruggedness must also be assessed and cannot be more than 20% RSD when the repeatability test is repeated on different days, on different instruments, or by different analysts. As with limit procedures, specificity must be demonstrated through unequivocal assessment of each target element (confirmation of the result using a secondary isotope).
The data in Table sVI (available online) show the summary results for the ICP-MS quant analysis of gelcaps, with six replicates for each spike level. The only data item not presented is the repeated analysis to demonstrate the ruggedness of the method, but a second analysis on a different day gave results well within the ruggedness criteria of no more than 20% RSD. Spike recoveries were all well within the required limits (70–150%) and repeatability was in the low % level, easily meeting the required acceptance criteria of no more than 20% RSD.


Table sVI: Accurate spike recovery and precision at 0.5J and 1.5J in gel cap samples (n = 6 for each sample)
Conclusions
The development of new USP <232> and <233> methodology for the preparation and analysis of pharmaceutical samples provides an opportunity for pharmaceutical laboratories to update their methodology and instrumentation and address the limitations of the current "heavy metals limit test" (USP <231>). With closed-vessel microwave digestion and element stabilization using HCl, ICP-MS has proven to be capable of routine determination of all regulated elements at low levels in pharmaceutical samples.
All regulated elements passed the acceptance criteria defined in USP <233> for both Limit and Quantitative procedures, including those elements that can suffer matrix-based interferences (such as the ArCl overlap on As), and those that are more soluble or stable in chloride matrix, particularly Hg which can be stabilized using a low percentage level of HCl. Routine addition of HCl to stabilize samples is no longer a problem for ICP-MS methods, because the resulting Cl-based polyatomic interferences are reliably removed using He cell gas mode on CRC-based ICP-MS instruments.
System performance validation data was easily within the method requirements for accuracy, precision, and spike recovery, and detection limits were all several orders of magnitude lower than the levels at which the trace elements are controlled.
Unequivocal identification and quantification of target elements is provided using secondary or qualifier isotopes for confirmation of the result reported at the primary isotope. ICP-MS also can be linked to a chromatography system to provide integrated separation and analysis of the different forms or species of As and Hg, as required under USP <232>.
Samina Hussain is with Exova USA in Santa Fe Springs, California.
Amir Liba and Ed McCurdy are with Agilent Technologies Inc in Santa Clara, California. Direct correspondence to: ed_mccurdy@agilent.com.
References
(1) K.B. Blake, Pharm. Forum 21(6), 1632–1637 (1995).
(2) R. Ciciarelli, D. Jäkel, E. König, R. Müller-Käfer, M. Röck, M. Thevenin, and H. Ludwig, Pharm. Forum 21(6), 1638–1640 (1995).
(3) N. Lewen, S. Mathew, M. Schenkenberger, and T. Raglione, J. Pharm. Biomed. Anal. 35(4), 739–752 (2004).
(4) T. Wang, J. Wu, R. Hartman, X. Jia, and R.S. Egan, J. Pharm. Biomed. Anal. 23(5), 867–890 (2000).
(5) "Elemental Impurities–Limits," Pharm. Forum 36(1), Chapter <232> (2011).
(6) "Elemental Impurities–Procedures," Pharm. Forum 36(1), Chapter <233> (2011).
(7) A.S. Al-Ammar and J. Northington, J. Anal. At. Spectrom. 26, 1531–1533 (2011).
(8) J. Huang, X. Hu, J. Zhang, K. Li, Y. Yan, and X. Xu, J. Pharm. Biomed. Anal. 40, 228–233 (2005).
(9) S. Lira, P. Brush, L. Senak, C. Wu, and E. Malawer, Pharm. Forum 34(6), 1613–1618 (2008)
(10) S.E. Jackson, B.J. Fryer, W. Gosse, D.C. Healey, H.P. Longerich, and D.F. Strong, Chem. Geol. 83, 119–132 (1990).
(11) A. Frimpong, B.J. Fryer, H.P. Longerich, Z. Chen, and S.E. Jackson, Analyst 120, 1675–1680 (1995).
(12) E. McCurdy and G. Woods, J. Anal. At. Spectrom. 19, 607–615 (2004).
(13) S. Wilbur and E. McCurdy, Spectros. 25(5), 2–7 (2010).










High-Speed Laser MS for Precise, Prep-Free Environmental Particle Tracking
April 21st 2025Scientists at Oak Ridge National Laboratory have demonstrated that a fast, laser-based mass spectrometry method—LA-ICP-TOF-MS—can accurately detect and identify airborne environmental particles, including toxic metal particles like ruthenium, without the need for complex sample preparation. The work offers a breakthrough in rapid, high-resolution analysis of environmental pollutants.




Trending on Spectroscopy: The Top Content of 2024
December 30th 2024In 2024, we launched multiple content series, covered major conferences, presented two awards, and continued our monthly Analytically Speaking episodes. Below, you'll find a selection of the most popular content from Spectroscopy over the past year.


