Demystifying SERS: A Newcomer’s Guide to Using Surface Enhanced Raman Scattering
SERS offers many advantages. However, there are several issues to be aware of when trying to use SERS signals in analytical applications.
It is fairly easy to generate a surface enhanced Raman scattering (SERS) signal. However, there are severalissues to be aware of when trying to use these signals in analytical applications. The SERS signal arises from the combination of the number of molecules (N), the polarizability or cross section of the molecule (σ), and the electric field (E) experienced by the molecules. Understanding how these different variables interact to generate the enhanced SERS response is the key to applying SERS accurately. Here, we try to share experiences and some helpful references to assist people interested in applications using SERS.
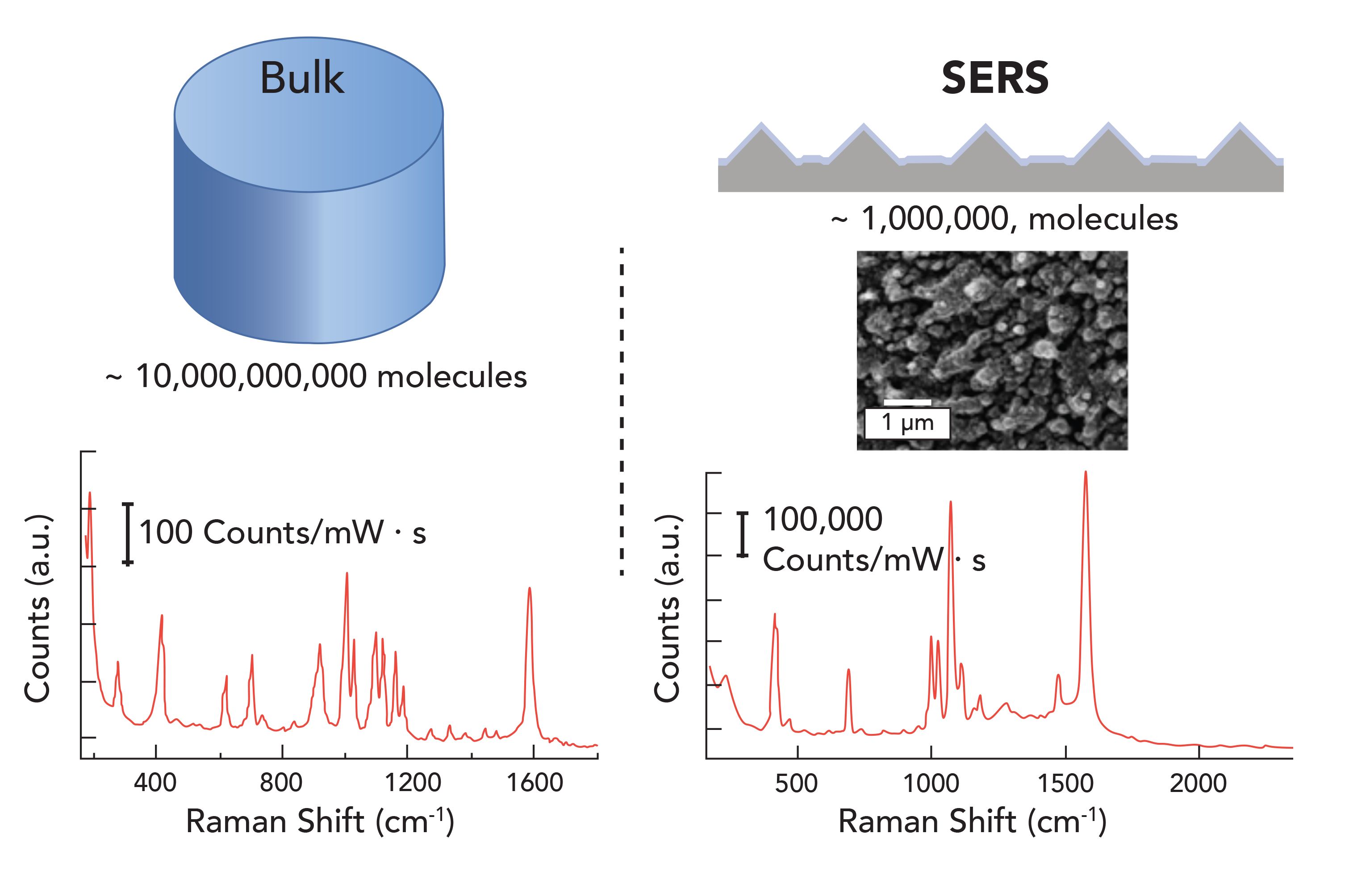
It has been more than40 years since the realization that Raman signals are enhanced on nanostructured metal surfaces (1). Figure 1 illustrates the advantages of surface enhanced Raman scattering (SERS), which provides a larger signal from fewer molecules. Over those past 40 years, significant progress has been made toward understanding the origin of the signal enhancement, enabling SERS to become a promising technique for chemical-specific analytical analysis. It is now understood how the electric fields originating from localized surface plasmon resonances on nanoparticles can concentrate the incident laser and generate significant enhancements in Raman scattering from molecules at the surface (2). In particular, nanoparticle dimers and larger aggregates with nanometer-scale gaps and crevices produce some of the largest electromagnetic field enhancements (3). In fact, it is fairly easy to generate a SERS signal; however, as many SERS practitioners will testify, there are a few things to be aware of when trying to use these signals in analytical applications (Table I). The SERS signal arises from the combination of the number of molecules (N), the polarizability or cross section of the molecule (σ), and the electric field (E) experienced by the molecules. Understanding how these different variables combine to generate the SERS response is the key to applying SERS. Here, I share my experiences and try to provide some helpful references to help people interested in applications using SERS. The following sections highlight some key concepts that people should be aware of when using SERS.


Figure 1: Illustration of how SERS generates a larger signal from fewer molecules. In the figure, the spontaneous Raman spectrum of thiophenol from a defined volume is compared to the SERS signal from an estimated number of thiophenol molecules on a nanostructured surface.



Not All Molecules Are Enhanced Equally
Much of the pioneering work in SERS has been performed using a molecule such as rhodamine or other molecules that have electronic resonance in the visible region (2). These experiments are often called surface enhanced resonance Raman scattering (SERRS). Just as resonance Raman increases the cross-section (σ) of spontaneous Raman scattering, it also enables the lowest limits of detection in SERS experiments. Most people studying single molecules use this additional enhancement (4). It turns out that many of these molecules are fluorescent dyes, and even with excitation away from the peak absorption, some level of pre-resonance is accessed boosting the SERS response.
The other class of molecules that is commonly detected in SERS is aromatic thiols and pyridines. Although these molecules do not have an intrinsic resonance, they are reported to form charge transfer complexes on the plasmonic surfaces that also increase the relevant cross-section of the analyte and boost the enhancement. These charge-transfer mechanisms are commonly associated with “chemical enhancement” (which has become a default category for enhancement that is not from the enhanced electric field).
Some molecules are just difficult. Glucose is one example. Although glucose has been detected by SERS, this often involves some form of surface functionalization or capture, such as reacting with boronic acid on the surface (5,6). It is often worth measuring the spontaneous Raman limit of detection for a molecule; with SERS one should be able to detect the molecule at a lower concentration. This is one way to distinguish SERS from weak spontaneous Raman scattering. However, if molar concentrations are needed to see the spontaneous Raman signal, the SERS signal may be difficult to measure.
SERS Enhances the Signal from the Molecules at the Surface
The SERS effect is a very short-range enhancement. Studies done on the distance dependence of SERS show the signal enhancement goes away within a few nanometers (7,8). Other studies indicate the molecule is physisorbed to the surface (9). There was even discussion of a “first layer effect,” where the molecules adsorbed to the surface showed a larger enhancement (10).The number (N) of molecules at the surface can often be modeled with a Langmuir isotherm, which correlates the surface coverage to the affinity of the molecule to the surface. At concentrations where your molecule is unlikely to have significant coverage, it is unlikely that one will detect SERS. Both electromagnetic and chemical enhancement mechanisms increase the signal of molecules at the surface. For nanoparticles dropped onto a substrate and dried, the molecules that stick to the nanoparticles are easier to detect.
The idea of a SERS tag is to put a molecule on the surface that gives a strong signal (see above) and then incorporate some form of chemical recognition agent (such as DNA, an antibody, or an aptamer) that binds the target molecule (11,12). Although these approaches have been very successful, it is worth remembering that your limit of detection and sensitivity will now be controlled by the chemical recognition agent on your tag.
Intensities Depend on the Nanostructures, Too
It has been shown that the large majority of the SERS signal originates from “hotspots,” (13) generally regarded as the high electric-field (E) regions associated with gaps and crevices. This means that one molecule in an electric field enhanced region of 106 will be as bright as either 106 molecules in solution, or 105 molecules in a region with an electric field enhancement of 10. This tells us that small changes in the number of molecules in “hotspots” can create large intensity variations. This is particularly problematic when using colloidal nanoparticles, where it can be very challenging to aggregate nanoparticles in a reproducible manner.
A lot of effort has gone into making patterned nanostructures as SERS substrates (14). Many of these substrate approaches still have some variation in the electric field enhancements, and it is common to observe intensity variation on the scale of 10% in these systems. One approach to averaging out this heterogeneity is to measure multiple spots. One study suggested that more than 100 spots are needed to properly capture this variance (15).
This is not to say that SERS cannot be quantitative (16). The use of internal standards, assumed to have the same intensity distribution as an analyte, can correct for this variance. This correction can be a co-adsorbed molecule (17), or, perhaps preferably, a stable isotope variant of the target molecule (18).
Enhancement Factor Madness
The metric used to compare the enhancement observed from a SERS active material is the enhancement factor (EF) (19). In theory, this number represents the increase in the Raman signal expected from a molecule. In practice, this is really difficult to determine. It requires that you measure a SERS signal (ISERS) from a known number of molecules (NSERS), and you have the spontaneous Raman signal (IRaman) from a known number of molecules (NRaman) under similar experimental conditions. With these four numbers, the EF can be calculated as follows:
[1]


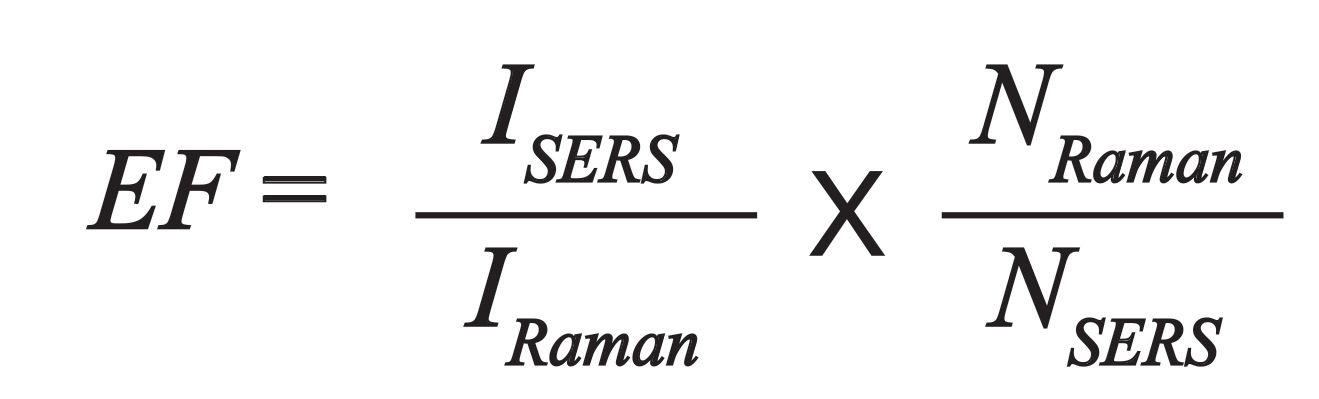
In perhaps the simplest case, a monolayer of thiol on a patterned nanostructured surface, approximations are still required. Because of the influence of hotspots, the number of molecules detected by SERS (NSERS) is difficult to know. Further, the free molecule may have a different cross-section than the adsorbed molecule, making IRaman also uncertain. Given these uncertainties, the EF is at best a relative number for the conditions under which it was determined. When reporting EFs, it is important to provide as much information as possible about how the EF was determined. As noted above, just because one molecule enhances well, another type of molecule may have a very different EF on the same nanostructures.
The Molecule You Put in Your Sample May Not Be What You Detect
One frustrating aspect of SERS is the vibrational frequencies observed may vary from those of the molecules in solution or powder forms, such as is observed in Figure 1. In SERS, there is a strong polarization dependence that selectively enhances those modes aligned with the enhanced electric field. In the past 10 years, experiments show that the electrons in the metals, which generate the plasmon resonance and enhance the Raman signal, can also do chemistry on the analytes. One classic example is para-aminothiophenol, where theories on changes in selection rules to activate Raman-inactive B2 modes were developed, only to find out the new frequencies arose from the formation of dimercaptoazobenzene on the surface (20). Understanding these effects is an active area of SERS research today. To avoid these effects, it is important to use low laser powers, generally less than 1 mW of power, in a diffraction-limited focus of a microscope objective focus. These low powers can minimize these photoreactions as well as heating that can cause problems in your measurement. For those seeking to use SERS, the best practice is to generate a calibration curve with known concentrations of your analyte at low laser powers, and use a strong band (or multivariate model) that provides the most accurate concentration determination.
References
- D.L. Jeanmaire and R.P. Van Duyne, J. Electroanal. Chem. 84, 1–20 (1977).
- P.L. Stiles, J.A. Dieringer, N.C. Shah, and R.R. Van Duyne, Annu. Rev. Anal. Chem. 1, 601–626 (2008).
- K.L. Wustholz, A.-I. Henry, J.M. McMahon, R.G. Freeman, N. Valley, M.E. Piotti, M.J. Natan, G.C. Schatz,and R.P. Van Duyne, J. Am. Chem. Soc. 132, 10903–10910 (2012).
- P.G. Etchegoin and E.C. Le Ru, Phys. Chem. Chem. Phys. 10, 6079–6089 (2008).
- B. Sharma, P. Bugga, L.R. Madison, A.I. Henry, M.G. Blaber, N.G. Greeneltch, N.H. Chiang, M. Mrksich, G.C. Schatz, and R.P. Van Duyne,J. Am. Chem. Soc. 138, 13952–13959 (2016).
- X. Gu, H. Wang, Z.D. Schultz, and J.P. Camden, Anal. Chem. 88, 7191–7197 (2016).
- J.A. Dieringer, A.D.McFarland, N.C. Shah, D.A. Stuart, A.V. Whitney, C.R. Yonzon, M.A. Young, X. Zhang, and R.P. Van Duyne, Faraday Discuss.132, 9–26 (2006).
- S. Lal, N.K. Grady, G.P. Goodrich, and N.J. Halas, Nano. Lett. 6, 2338–2343 (2006).
- S.M. Asiala, and Z.D. Schultz, Anal. Chem. 86, 2625–2632 (2014).
- A. Otto, Phys. Status Solidi. A 188, 1455–1470 (2001).
- D. Graham, B.J. Mallinder, and W.E. Smith, Angew. Chem., Int. Ed. 39, 1061–1063 (2000).
- N.R. Isola, D.L. Stokes, and T. Vo-Dinh, Anal. Chem. 70, 1352–1356 (1998).
- Y. Fang, N.-H. Seong, and D.D. Dlott, Science 321, 388–392 (2008).
- B. Sharma, M.F. Cardinal, S.L. Kleinman, N.G. Greeneltch, R.R. Frontiera, M.G. Blaber, G.C. Schatz, and R.P. Van Duyne, MRS Bull. 38, 615–624 (2013).
- S. Mabbott, Y.Xu, and R. Goodacre, Anal. Meth. 9, 4783–4789 (2017).
- S.E.J. Bell and N.M.S. Sirimuthu, Chem. Soc. Rev. 37, 1012–1024 (2008).
- A. Nguyen and Z.D. Schultz, Analyst 141, 3630–3635 (2016).
- A. Subaihi, Y. Xu, H. Muhamadali, S.T. Mutter, E.W. Blanch, D.I. Ellis, and R. Goodacre, Anal. Meth. 9, 6636–6644 (2017).
- P.L. Stiles, J.A. Dieringer, N.C. Shah, and R.P. Van Duyne, Annu. Rev. Anal. Chem. 1, 601–626 (2008).
- Y.-F. Huang, H.-P. Zhu, G.-K.Liu, D.-Y. Wu, B. Ren, and Z.-Q. Tian, J. Am. Chem. Soc.132, 9244–9246 (2010).
Zachary D. Schultz is with the Department of Chemistry and Biochemistry at The Ohio State University, in Columbus, Ohio. Direct correspondence to:
Schultz.133@osu.edu











AI-Powered SERS Spectroscopy Breakthrough Boosts Safety of Medicinal Food Products
April 16th 2025A new deep learning-enhanced spectroscopic platform—SERSome—developed by researchers in China and Finland, identifies medicinal and edible homologs (MEHs) with 98% accuracy. This innovation could revolutionize safety and quality control in the growing MEH market.


New Raman Spectroscopy Method Enhances Real-Time Monitoring Across Fermentation Processes
April 15th 2025Researchers at Delft University of Technology have developed a novel method using single compound spectra to enhance the transferability and accuracy of Raman spectroscopy models for real-time fermentation monitoring.

Nanometer-Scale Studies Using Tip Enhanced Raman Spectroscopy
February 8th 2013Volker Deckert, the winner of the 2013 Charles Mann Award, is advancing the use of tip enhanced Raman spectroscopy (TERS) to push the lateral resolution of vibrational spectroscopy well below the Abbe limit, to achieve single-molecule sensitivity. Because the tip can be moved with sub-nanometer precision, structural information with unmatched spatial resolution can be achieved without the need of specific labels.



