Mass Cytometry: An Evolution in ICP-MS Enabling Novel Insights in Single-Cell Biology
A novel adaptation to inductively coupled plasma–mass spectroscopy (ICP-MS), mass cytometry provides researchers with a tool to study the complexity of biology at the single-cell level.
A novel adaptation to inductively coupled plasma–mass spectroscopy (ICP-MS), mass cytometry provides researchers with a tool to study the complexity of biology at the single-cell level. New insights into the interrogation of high dimensional data systems using unsupervised data networking algorithms are enabling the application of mass cytometry to the study of the diversity of cell subsets, as well as to determine cellular functions such as apoptosis, cell signaling pathways, the cell cycle, and DNA damage.
Mass cytometry uses reporter molecules (typically antibodies) tagged with transition-metal isotopes to label cells from blood, tissues, or cell cultures. The metal-tagged antibodies specifically bind to their target proteins, and the mass cytometer measures the expression of these biomarkers in each individual cell according to the type and amount of each metal detected. The current instrument configuration provides users with more than 120 detection channels at an acquisition rate of up to 1000 cells per second.
Mass cytometry has been used as a tool for accurate cell profiling, providing researchers with a detailed understanding of the diversity of cell subsets, as well as to determine cellular functions such as apoptosis, cell signaling pathways, the cell cycle, and DNA damage. Using levels of expression of particular cell differentiation markers, researchers have dramatically improved the ability to discern the cell lineages and cellular hierarchy in hematopoiesis. Bendall and colleagues (1) recently used a 44-marker phenotyping and signaling panel to discover and functionally profile rare B cell progenitors involved in immunoglobulin heavy chain rearrangement and to order cells according to their developmental timing. The intensity of expression of these cell lineage markers provides insight into the unique cell differentiation pathway of hematopoietic cells and detects multiple pathways across a number of cell subsets (2,3).
The mass cytometer provides researchers with a tool to examine normal and diseased cells and interrogate the cells to understand biological processes. The addition of cytokines and drugs to stimulate or suppress specific markers provides a more complete picture of the pathway-specific signaling responses in various cell types (3). Cancer-specific panels of markers are designed to provide a deeper understanding of the intracellular biology involved in the progression of cancers and drug response (including resistance) and can be expected to lead to more-informed treatment decisions in the future.
Isotope Resolution
Mass cytometry provides an advantage over current flow cytometry methods by obviating the need for fluorescence compensation because of spectral overlap of fluorochromes. The resolution of the mass cytometer allows discrimination of isotopes separated by one atomic mass unit with minimal spectral overlap and increases the number of markers that can be measured simultaneously in a single cell, as seen in Figure 1.


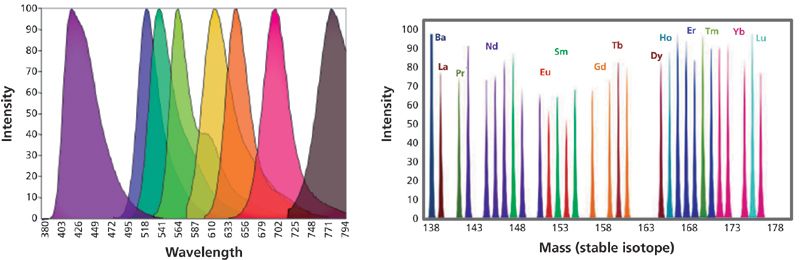
Figure 1: An eight-parameter flow cytometry experiment illustrating the spectral overlap between the fluorochromes. Mass cytometry involves the collection of mass spectral data in distinct channels with 1 amu space between each channel.
Currently, 15 transition metals provide up to 36 isotopes with unique masses that fall within the mass range of detection of the instrument as exemplified in Figure 2. The palladium (Pd) metal is also highlighted here because of its utility in cell barcoding using mass cytometry, which is discussed in detail below. These isotopes have low natural abundance in biological systems and therefore will not interfere with single-cell analysis (4).


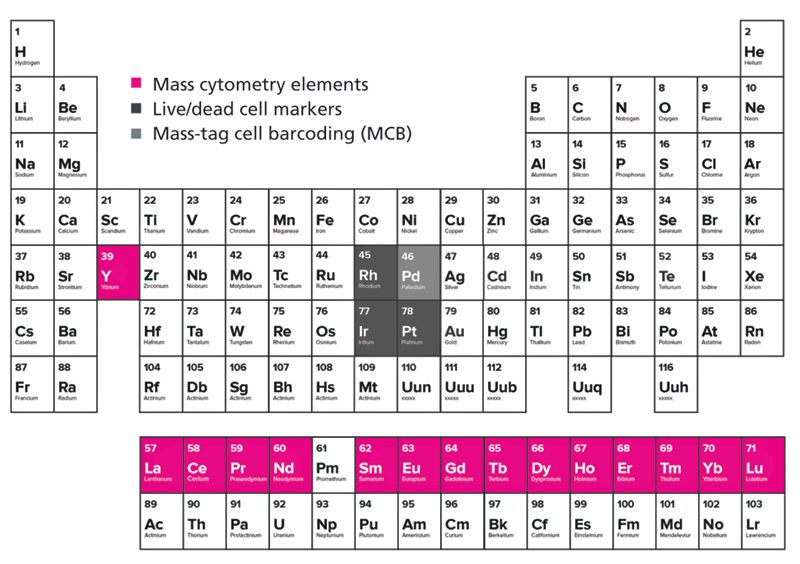
Figure 2: The periodic table highlighting the lanthanide-transition group of elements (pink), the live-dead cell markers (dark grey), and the mass tag cell barcoding metal (light gray) used in mass cytometry experiments. Rh, Ir, and Pt are used for the determination of cell viability in mass cytometry, and Pd isotopes can be used for mass tag cell barcoding (MCB).
The reporter metal tags are attached to antibodies through the use of metal-chelating polymer (MCP) labels. MCPs are water-soluble polymers bearing multiple metal-chelating ligands and are critical components of mass cytometric bioassays. These metal-chelating polymers incorporate 1,4,7,10-tetraazacylcododecane-1,4,7,10-tetraacetic acid (DOTA) or diethylene triamine pentaacetic acid (DTPA) (4,5), that bind the metal and a terminal maleimide group for coupling to the Fc portion of an antibody. The performance of the metal-chelating polymers in biological applications is related to their own structure. Current research is focused on the development of metal-chelating polymers with different structures or longer chains that will allow for the incorporation of more metal chelators, thereby increasing the signal levels of the metal reporter and extending the function of the mass cytometer instrument. The other expanding area of research involves the use of water-soluble lanthanide up-conversion nanoparticles, which can increase the number of metal atoms incorporated to provide a stronger signal for low copy number biomarkers. The surface functionalization and bioconjugation of these molecules has been the topic of intense research in recent years.


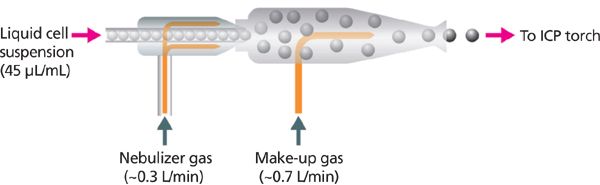
Figure 3: The cell suspension is injected into the nebulizer as argon gas flows through the nebulizer and spray chamber, carrying the sample to the ICP torch. A makeup gas is used to optimize the plasma conditions independently of the optimization of the "nebulizer gas," which impacts the integrity of individual cells.
Instrumental Components
The mass cytometer technology (CyTOF 2, Fluidigm Corporation) discussed in this column installment consists of five major components including sample introduction, the inductively coupled plasma (ICP), the ion optics and time-of-flight (TOF) analyzer, and the detector. Sample in the form of a liquid cell suspension is introduced into the mass cytometer instrument at a flow rate of 45 μL/min within a stream of argon gas as shown in Figure 3. The cells pass through a concentric nebulizer and exit through a tapered end, where they are aerosolized into fine single-cell droplets, after which they pass into the spray chamber. The spray chamber is heated to a temperature of approximately 200 °C, resulting in the vaporization of the remaining water associated with the cells within the heated argon gas, minimizing sample condensation as it travels toward the plasma. The result is that individual cells exit the spray chamber in a humid argon gas stream. The complete list of operating conditions are shown in Table I.


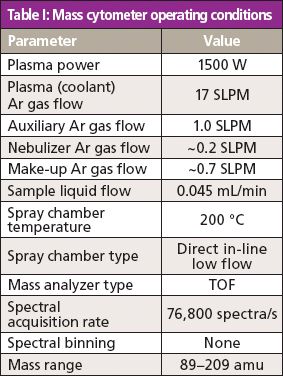
Table I: Mass cytometer operating conditions
Principles of Operation
The single-cells associated with the specific isotope-tagged antibodies are injected into the ICP and subsequently vaporized, atomized, and ionized.
Figure 4 depicts the ICP and the formation of ion clouds from individual cells stained with metal-labeled antibodies. The particles in the sample that are introduced into the plasma are ionized to produce an ion cloud. The ion cloud associated with individual particles, which includes the ionized metal tags that report on the protein content of the cells, diffuses as it flows in the plasma. At the point of extraction, the ion cloud is approximately 2 mm in diameter and is flowing at a velocity of approximately 10 m/s. The plasma is extracted through a three-aperture plasma–vacuum interface, such that the ion cloud produces a transient pulse of cell-associated particles that is pseudo-Gaussian with a half width of ~200 μs. After the ions are largely separated from the un-ionized plasma gas, they pass through a high pass optic that transmits ions above ~80 amu that are then directed toward the TOF analyzer, wherein they are separated according to their mass-to-charge ratio. The signal from the detector is digitized by an analog-to-digital converter (ADC) and the data in time are converted to mass and integrated into an analog intensity value and an integrated number of ions (4). The mass spectra are recorded in 13-μs intervals, and the data are exported as FCS3.0 files, which are considered the standard flow cytometry data file format (6).


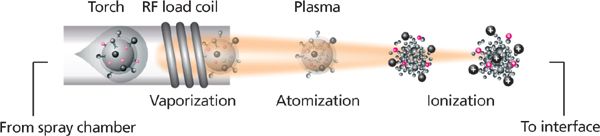
Figure 4: The vaporization, atomization, and ionization of cell droplets in the high-temperature argon plasma. Cells are extracted through a vacuum interface on their journey to the TOF mass analyzer.
Because of the high dimensionality and complexity of the data collected, several analytical techniques have been applied to mass cytometry, some examples of which include spanning tree progression analysis (SPADE), principal component analysis (PCA), and the t-distributed stochastic neighbor embedding (t-SNE) algorithm called viSNE. These unsupervised data analysis tools are discussed in further detail.
Mass Cytometry Workflow
The workflow of mass cytometry parallels that of conventional (optical) flow cytometry. Various monoclonal and polyclonal antibodies reactive with intracellular and cell surface antigens that are validated for flow cytometry can be used to recognize their corresponding proteins, which is demonstrated in Figure 5. A solution of enriched lanthanide is added to the polymer and incubated, followed by several washing steps. The antibody is conjugated to the metal-loaded polymer. For intracellular marker staining, cells are fixed and permeabilized, then incubated with the metal-tagged antibodies (4). The efficiency and avidity of tagging antibodies is improved with large-scale conjugation and is, in part, responsible for the trend toward using commercially available metal-labeled reagents.


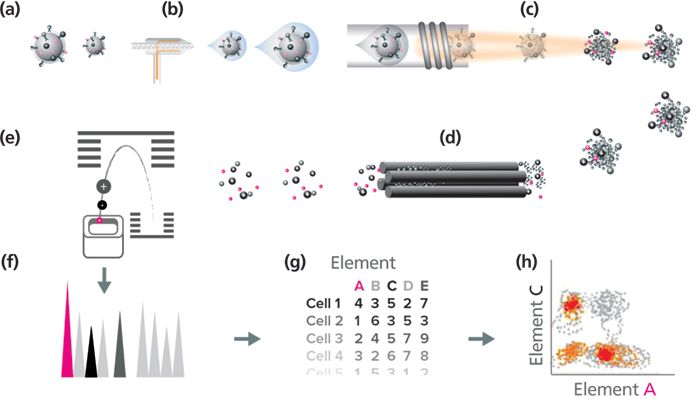
Figure 5: Mass cytometry workflow: (a) Cells labeled with metal-conjugated antibodies in solution are injected into (b) the nebulizer. They are aerosolized and reduced to single cell-containing droplets. (c) The cells are directed to the ICP torch, where they are vaporized, atomized, and ionized in the plasma. (d) The high pass optic removes the low-mass ions and the photons, resulting in an ion cloud that enters (e) the TOF mass analyzer. The ions are separated based on their mass-to-charge ratio and are accelerated to the detector. (f) The detector measures the quantity of each isotope for each individual cell in the sample; data are generated in (g) an FCS format and (h) analyzed.
Methods for the Determination of Viable Cells
Usually, one to three million cells are surface-stained with the metal-conjugated antibodies. The ability to determine the number of live or dead cells in an experiment is crucial to the dependability of data generated by mass cytometry. There are two currently accepted techniques used in mass cytometry. The first involves the addition of iridium(III) Ir- and rhodium(III) Rh-intercalators. The intercalators are used in nuclear staining and are stable in aqueous solution (4). Cells are first stained with Rh-intercalator, which stains only the dead cells, and then the Ir-intercalator, which is applied to fixed and permeabilized cells at the end of the staining protocol, thus staining all cells. This process allows for the determination of viable cells. This also provides a flag for cellular events that can be useful during data interpretation.
The second accepted method for determination of viable cells involves the use of cisplatin. Recent studies cite platinum-based compounds as beneficial in the identification of nonviable cells. For example, cisplatin, a DNA damaging agent used in chemotherapy, can be used to resolve live and dead cells using mass cytometry (7). In this case, commercially available cisplatin contains 195Pt. Analogous to the commercially available fluorescent viability dyes such as propidium iodide and 7-aminoactinomycin (7-AAD), cisplatin can penetrate the plasma membrane and react with proteins in dead cells. The other advantage of incorporating this compound into mass cytometry experiments compared to other viability reagents is that it is applicable to a wide range of mass cytometry experiments including suspension cells and primary tissue samples and the resulting covalent modifications ensures stability during cell processing, which may involve several cycles of washing (7).
Compensating for Signal Drift
A significant variable to consider in most ICP-mass spectrometry (MS) instruments is that they undergo some drift in the signal over time because of several different factors including a buildup of cellular material and daily maintenance procedures that can alter performance between samples. The reflection of these variations in the final data can be minimized using normalization algorithms built into the software, using a particle-based analog of internal standardization. Samples are spiked with commercially available metal-encoded polyacrylamide beads that incorporate four lanthanides - cerium, europium, holmium, and lutetium - encompassing the middle masses of the range of the instrument. Conveniently, the use of normalization beads allows not only normalization within a run or between different runs on the same instrument, but also allows direct comparison between different instruments and different laboratories.
Cell Barcoding in Mass Cytometry
Ideally, the conditions for sample preparation of mass cytometry samples in a given experiment across multiple stimulations should be uniform, therefore there is an advantage to processing samples and acquiring data from related samples simultaneously (6). One approach to improve the efficiency of multiparameter assays uses pooled sample analysis with cell barcoding and has been used extensively in various types of bioassays such as DNA sequencing and enzyme-linked immunosorbent assays (ELISA) (8). This technique, originally conceived as fluorescence cell barcoding, is now being applied to mass cytometry experiments. Mass-tag cell barcoding (MCB) relies on the addition of unique combinatorial barcodes of metal isotopes to label individual cell samples that are then pooled for processing and measured as one single multiplexed sample (9). Multiplexing of samples using MCB increases sample throughput, eliminates variability between samples during antibody tagging procedures, reduces consumption of reagents, and improves instrument sensitivity (8,9). Palladium's six naturally occurring isotopes - 102Pd, 104Pd, 105Pd, 106Pd, 108Pd, and 110Pd - make it an excellent candidate as a barcoding reagent. The use of these Pd-based barcoding reagents does not interfere with antibody detection channels that are in wide use now, and yet still falls within the mass range of the instrument. The barcoding scheme also helps to filter out doublets, or the false interpretation of two or more cells as a single cell (8). The singlet barcode is represented by the presence of only three of the six possible barcodes (6-choose-3 MCB). The scheme allows the multiplexing of up to 20 different samples using barcoding while eliminating the doublets for rapid unbiased sample deconvolution. Table II shows the barcode scheme as applied to mass cytometry.


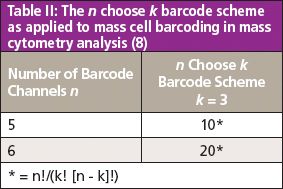
Table II: The n choose k barcode scheme as applied to mass cell barcoding in mass cytometry analysis (8)
Interpretation of Data
After the data have been collected and are ready for analysis there are several options for interpretation of mass cytometry data. Currently, two platforms are the most commonly used to support cytometry data: Cytobank (10) and FlowJo (11). Data can be imported into Cytobank, and several forms of data interpretation can be carried out within this platform. Cytobank provides users the ability to look at large datasets in parallel. Researchers can manipulate, analyze, and share these large datasets. FlowJo allows users to manipulate datasets in a variety of ways: gate the data and remove outliers, select from various types of graphs, and change individual parameters. Users can isolate single cell types from a population and then overlay bivariate two-dimensional (2D) scatter plots of combinations of all the parameters in the experiment.
Data collected from mass cytometry experiments contains extremely high dimensionality; therefore, supervised methods of data analysis may require significant involvement of the data analyst. There are several methods that researchers can use to interrogate data obtained in mass cytometry experiments that are unsupervised and hypothesis-generating (6). This type of computer-assisted gating and clustering of populations may provide users with a better view of the bigger picture. Let's review three of the types of software used in recent publications: PCA, SPADE, and viSNE.
PCA takes data collected with a large number of variables for a single population and reduces it to a few interpretable linear combinations. This condenses the dimensionality of the dataset to provide a low-dimensional picture. This method was used by Newell and colleagues (12). Metal-encoded tetramer probes were used to stain human peripheral blood samples and these were examined using 25 metal-labeled antibodies to cell differentiation and signaling molecules. The subpopulations of CD8+ memory T-cells were gated by known surface marker phenotypes in CD8+ memory T-cells in human peripheral blood samples. Data from subpopulations of cells was clustered according to phenotypic and functional characteristics. The results of the study illustrated the diversity of memory cells and the changes in expression levels of markers as the cells transition from effector memory cells to central memory cells.
Another example of algorithms that cluster larger cohorts of samples, SPADE, provides a platform to cluster data in multidimensional space and reduce it to a 2D representation using the minimum spanning tree algorithm. This methodology provides a means to extract hierarchical data from high-dimensional mass cytometry analysis in an unsupervised manner. The tool has been integrated into both FlowJo and Cytobank and provides the ability to interrogate various parameters between clusters. A transformational study by Bendall and colleagues (2) compared 34 parameters simultaneously in healthy human bone marrow samples using single-cell mass cytometry. The study examined 18 intracellular proteins and 13 cell surface markers and the response to various immune modulators and drugs. The data was compared to results obtained by flow cytometry. The large bubbles in Figure 6 (15) represent the cell populations based on surface marker expression (manually annotated), and the size of each circle within the bubble represents the relative frequency of cells that fall within a 13-dimensional node. The study provided a thorough view of the continuum of cellular differentiation within cell subsets in the human hematopoietic system based on surface antigen expression, and a deeper understanding of cell and humoral immunity.


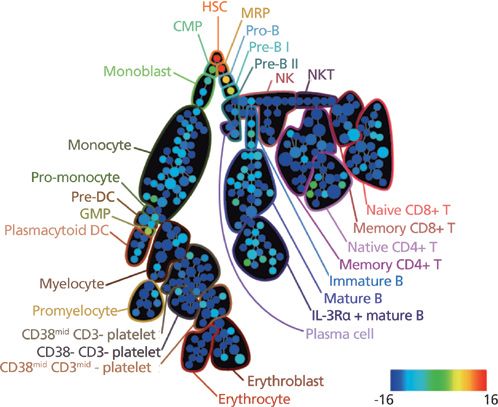
Figure 6: The SPADE analysis of human hematopoietic cells in human bone marrow samples. CD34 (shown here) was used to define the T-cell lineages. Adapted from reference 15 with permission from Elsevier.
A third type of clustering tool used extensively in mass cytometry is viSNE, which is used to reduce high-parameter data to two dimensions, providing a visual picture of rare biological subsets, and gate single-cell events across different samples (13). Cells are placed on a 2D biaxial plot, with related cells being positioned closer together than unrelated cells. Recent studies examined healthy human bone marrow cells and compared this to bone marrow in leukemic patient samples using viSNE (14). The maps generated by these two populations had a very distinct display in the 2D space, and the leukemia samples were less consistent in shape compared to the healthy cells (14). Other studies have looked at Tregs (regulatory T cells) from peripheral blood mononuclear cells (PBMCs) and have confirmed that the presence of naïve, effector, terminal effector, and Helios subsets of Tregs can be determined by examining a panel of nine cell surface and intracellular markers using viSNE analysis (13).
Final Thoughts
Mass cytometry builds on the capabilities of ICP-MS to exquisitely detect and provide absolute quantification of stable elements. The coupling of antibodies to cell surface or intracellular antigens to metal-containing polymers allows the detection of a multitude of proteins in the cell. The resulting high dimensional datasets can be clustered using various data mining algorithms that are unsupervised in nature, thereby providing researchers with critical insights into the relationships between proteins in the cell. Atomic mass spectrometry has already provided important insights into disciplines as diverse as immunology, cancer biology, vaccine development, drug discovery, and translational medicine. The emerging recognition of the importance of studying biology at the single cell level, and the depth of information that simultaneous determination of many proteins offers, is reflected in the number and breadth of application publications using mass cytometry. We should anticipate that the development of novel MCPs will expand the assay panel to well beyond 36 simultaneous parameters, and that creative approaches to data analysis will expose the complexity of cellular systems and thus improve our understanding of disease, health, and the transition between them.
References
(1) S.C. Bendall, K.L. Davis, E.-a.D. Amir, M.D. Tadmor, E.F. Simonds, T.J. Chen, D.K. Shenfeld, G.P. Nolan, and D. Pe'er, Cell157(3), 714–725 (2014).
(2) S.C. Bendall et al., Science332, 687–696 (2011).
(3) S.C. Bendall and G.P. Nolan, Nat. Biotechnol.30, 639–647 (2012).
(4) O. Ornatsky, D. Bandura, V. Baranov, M. Nitz, M.A. Winnik, and S. Tanner, J. Immunol. Methods361, 1–20 (2010).
(5) X. Lou, G. Zhang, I. Herrera, R. Kinach, O. Ornatsky, V. Baranov, M. Nitz, and M.A. Winnik, Angew. Chem. Int. Ed. Engl. 46, 6111–6114 (2007).
(6) S.D. Tanner, V.I. Baranov, O.I. Ornatsky, D.R. Bandura, and T.C. George, Cancer Immunol. Immunother.62, 955–965 (2013).
(7) H.G. Fienberg, E.F. Simonds, W.J. Fantl, G.P. Nolan, and B. Bodenmiller, Cytometry A81, 467–475 (2012).
(8) E.R. Zunder, R. Finck, G.K. Behbehani, E.-a.D. Amir, S. Krishnaswamy, V.D. Gonzalez, C.G. Lorang, Z. Bjornson, M.H. Spitzer, B. Bodenmiller, W.J.l. Fant, D. Pe'er, and G.P. Nolan, Nat. Protoc.10, 316–333 (2015).
(9) B. Bodenmiller et al., Nat. Biotechnol.30, 858–867 (2012).
(10) Cytobank, available at: https://www.cytobank.org/.
(11) FlowJo Software (Version 10), available at: http://www.flowjo.com/.
(12) E.W. Newell, N. Sigal, N. Nair, B.A. Kidd, H.B. Greenberg, and M.M. Davis, Nat. Biotechnol. 31, 623–629 (2013).
(13) M.A.P. Konrad, E.F. Simonds, S.C. Bendall, T.C. George, T.J. Chen, and P. Delucci, Analysis of Mass Cytometry Data with viSNE Reveals Phenotypic Heterogeneity with Human Tregs (American Academy of Immunology, 2014).
(14) E.-a.D. Amir, K.L. Davis, M.D. Tadmor, E.F. Simonds, J.H. Levine, S.C. Bendall, D.K. Shenfeld, S. Krishnaswamy, G.P. Nolan, and D. Pe'er, Nat. Biotechnol.31(6), 545–552 (2013).
(15) G.P. Nolan in Best Practice & Research Clinical Haematology24(4) (Elsevier Ltd., 2011), pp. 505-508, http://www.sciencedirect.com/science/article/pii/S1521692611000855.
Farah Virani, MSc, is a scientific and technical writer at Fluidigm Corporation in South San Francisco, California.


Farah Virani
Scott Tanner, PhD, is one of the four inventors of the mass cytometer and is currently the Chief Technology Officer (CTO) at Fluidigm Corporation. Direct correspondence to: Scott.Tanner@fluidigm.com



Scott Tanner






LIBS Illuminates the Hidden Health Risks of Indoor Welding and Soldering
April 23rd 2025A new dual-spectroscopy approach reveals real-time pollution threats in indoor workspaces. Chinese researchers have pioneered the use of laser-induced breakdown spectroscopy (LIBS) and aerosol mass spectrometry to uncover and monitor harmful heavy metal and dust emissions from soldering and welding in real-time. These complementary tools offer a fast, accurate means to evaluate air quality threats in industrial and indoor environments—where people spend most of their time.





Atomic Perspectives: Highlights from Recent Columns
March 3rd 2025“Atomic Perspectives,” provides tutorials and updates on new analytical atomic spectroscopy techniques in a broad range of applications, including environmental analysis, food and beverage analysis, and space exploration, to name a few. Here, we present a compilation of some of the most popular columns.

