A Mathematical Approach to Error Reduction in Mass Spectrometry
Special Issues
The exploding field of proteomics has highlighted the need to improve the performance of mass spectrometry, both quantitatively and qualitatively. These needs have led instrument manufacturers to produce instruments of increasingly higher quality, but little work has been done to address the fundamental errors inherent in the measurement technique itself. This paper examines these errors and demonstrates that the appropriate mathematical correction of MS data can significantly improve the performance of both low- and high-resolution instruments.
The dependence of mass accuracy (expressed as standard error σ in ppm) upon signal strength (S) and mass spectral resolving power (R) has been reported in the literature (4, 5) as:


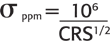
where the constant C includes such factors as signal unit conversion to real ion counts, peak sampling interval, peak analysis, and mass determination algorithms.
Reflecting on this equation, two important conclusions can be drawn:
- Mass accuracy is proportional to the square root of the signal strength; and
- C is dependent upon the data analysis methods outside of the physical measurement process.
The first conclusion indicates that even for low-resolution instruments, we should see a significant gain in mass accuracy for measurements with high signal-to-noise ratios (S/N). This should be particularly useful in low-resolution instruments, which tend to have increased ion transmitting efficiency. This means we should not see a significant loss in mass accuracy in instruments of low resolving power due to their intrinsically higher ion transmitting efficiency (or an effectively larger aperture, to borrow a term from optical spectroscopy).
The second factor involves the mysterious constant C. As defined above, it is clear that many of the parameters relating to C also lie outside the measurement process. This provides the opportunity to minimize the errors by way of calibration and analysis methods. Interestingly, published values for C rarely have been calculated for commercial instruments with the exception of the article mentioned above. By refining the analysis process, it should be possible to reduce the error for both high- and low-resolution instruments.



Table I. A brief summary of improvements in Mass Accuracy (MA) using HAMSCA.
Methodology
Based upon this simple analysis of errors, the authors proceeded with an in-depth analysis of the current methods of peak analysis. Current methods can be summarized as follows:
- Run a calibration standard with multiple ions covering the mass spectral range of interest
- Locate ion peaks and calculate peak centroids (peak centroiding)
- Fit a calibration correction curve between the calculated peak centroids and known peak masses
- Run the sample, possibly including an internal standard
- Apply the calibration correction curve to the mass axis of acquired scan
- Adjust the calibration curve if necessary based on the internal standard
- Calculate the peak areas based upon assumed peak start and peak end parameters, and calculate peak center masses through another layer of peak centroid calculation (peak centroiding) with assumed peak definition parameters.
This approach suffers from noise and baseline interferences for low signals, asymmetrical peak shapes for high signals, the changes in peak shapes from time to time, isotope interferences from unit mass resolution systems, and the intrinsic errors in peak centroiding through the use of only part of the mass spectral peak data in the calculation.


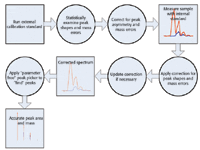
Figure 1. A simplified flow diagram for HAMSCA.
A new approach (referred to here as the highly accurate mass spectral calibration approach, or HAMSCA) addresses all these and other factors in mass spectral data analysis such that all systematic variations are calibrated out through the use of sufficient external and internal standards. No peak distortions or other artifacts are introduced by using a comprehensive mathematical data analysis process to achieve optimal signal processing and averaging (maximizing C in the above equation), and the quantitative accuracy (peak area integration) and qualitative accuracy (mass accuracy) are limited only by random errors in ion detection. This new method can be summarized as follows:
- Run a calibration standard with multiple ions covering the mass spectral range of interest
- Statistically examine peak shapes and mass errors for the calibration standard
- Build a correction to compensate for any mass errors and peak asymmetries
- Run the sample, possibly including an internal standard
- Apply the correction to compensate for both mass errors and peak shape asymmetries
- Update the correction if necessary based upon the internal standard
- Find peak areas and peak center masses using a parameter-free peak picker without the use of any assumptions.
Experimental Results
Test runs to validate HAMSCA were performed on a variety of instrument types from different vendors. Both low resolution (0.5-5 Da FWHM) and higher resolution instruments (~0.1 Da FWHM) were tested. First an instrument calibration standard is run on the instrument. The data is transferred to the computer program, which processes the data using HAMSCA. Key peaks are designated, and the software calculates the correction function for the spectrometer automatically. Next the sample is run in the normal fashion using the normal instrument vendor-provided software. An internal standard is included when seeking maximum mass accuracy. The sample spectrum then is transferred back to the program, which processes the raw spectrum to obtain the fully calibrated and aligned spectrum. Finally, a mathematically sound peak analysis algorithm is applied to the processed spectrum to yield high mass accuracy and unbiased peak picking results.
Figure 2 depicts the results obtained from a typical lower resolution instrument. An external standard of sodium trifluoroacetate (STFA) solution was used to develop the correction function. A sample of terfenadine was run along with internal standards of reserpine and promethazine. The monoisotopic mass was calculated to be 472.3191 versus the actual mass of 472.3216. This is about 5 ppm in mass accuracy as compared with the typical observed error of ~500 ppm in normal instrument operation. In addition, it should be noted that there is substantial enhancement in S/N (about 3X) as well as improvements in peak shape and symmetry which greatly enhances the ability to perform automated peak detection.


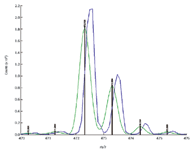
Figure 2. A mass accuracy of 5 ppm is achieved on Sciex Q Trap 4000 instrument of unit mass resolution for the drug compound terfenadine (exact monoisotopic mass 472.3216).
Figure 3 depicts the results from a higher resolution instrument with a typical observed mass accuracy of 5 ppm (four-scan average) using the vendor-provided software routines. In this case, both an internal and external standard was used in the calibration. The mass accuracy was improved in this case to 2.5 ppm when using HAMSCA. Again note the substantial improvement in peak shape and noise as well.


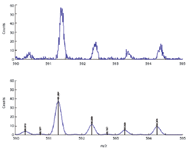
Figure 3. From top to bottom shows processing before and after HAMSCA. Sample: polyalanine solution; internal standard: 8-alanine ion (C24H43N8O9+, monoisotopic mass 587.3153); external standard: polyalanine standard; instrument: Waters Micromass qTOF II.
Figure 4 is an example of a low-resolution system run using only an external standard. While the best mass accuracy is obtained using internal standard, in this case it was not of major concern. It can be seen that the HAMSCA method of processing still offers substantial improvements in S/N for improved detection limits and peak shape for improved quantitative accuracy.


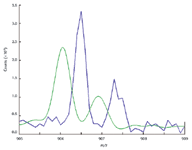
Figure 4. In addition to mass accuracy improvement, significant improvement in S/N is gained as well (3X or greater). Sample: sodium trifluoroacetate (STFA) solution; internal standard: none; external standard: STFA standard; instrument: Sciex Q Trap 4000.
To validate the operability of HAMSCA across different instrument designs and vendors, the authors have attempted to measure results on as large a cross section of available instruments as possible. Table I illustrates a subset of some of the results obtained.
Conclusion
It has been shown that current MS processing techniques typically fall far short of extracting the maximum amount of information from the data. Significant improvements in MS S/N and peak shape are obtained by simply running an external standard and using it as a reference to calculate a mathematically accurate correction function. With the addition of an internal standard, HAMSCA can routinely extend the capabilities of unit mass resolution instruments to obtain mass accuracies of up to 5 ppm. This is approximately 100X better than results typically obtained and suitable for accurate compound identification, a capability usually available only on more expensive systems. This might make it possible for relatively low-cost instruments to be used routinely for biomarker discovery, metabonomics, and proteomics research.
Substantial improvements also are seen on higher resolution spectrometers in S/N, peak shape, and mass accuracy. Although mass accuracy improvements are not as dramatic on higher end instruments, the improvements still are substantial and can improve the results in the above mentioned application areas. However, since these instruments typically are purchased to perform high mass accuracy measurements, the resulting improvements might be of greater value for users of these systems by further increasing the accuracy and confidence in the data.
Finally, due to the fact that the algorithms used are linear operations, the speed of processing could allow the results to be calculated in real-time on vendor supplied software.
References
1. M.P. Balogh,
LCGC Eur.
17
(3), 152 (2004).
2. K.L. Busch, Spectroscopy 19 (11), 32 (2004).
3. K.L. Busch, Spectroscopy 17 (10), 32 (2002).
4. K.F. Blom, Anal. Chem. 73, 715 (2001).
5. Moini et al., JASMS 9, 977 (1998). ?
Ming Gu,Yongdong Wang, and Don Kuehl are with Cerno Bioscience (New Haven, CT; www.cernobioscience.com)














. Photo Credit: © Kelsey Williams.")


High-Speed Laser MS for Precise, Prep-Free Environmental Particle Tracking
April 21st 2025Scientists at Oak Ridge National Laboratory have demonstrated that a fast, laser-based mass spectrometry method—LA-ICP-TOF-MS—can accurately detect and identify airborne environmental particles, including toxic metal particles like ruthenium, without the need for complex sample preparation. The work offers a breakthrough in rapid, high-resolution analysis of environmental pollutants.



