Spectroscopy Is Applied Quantum Mechanics, Part IV: Ideal Systems
In Parts I-III of this series, columnist David W. Ball recounted the failings of classical mechanics, the quantum hypothesis, and the rise of a new theory called quantum mechanics. In this installment, he discusses the ideal systems whose wavefunctions can be determined exactly from the Schr?dinger equation.



In the last installment, we introduced the one-dimensional Schrödinger equation, a central expression in quantum mechanics:


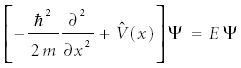
Here, h is Planck's constant divided by 2π, m is the mass of the material in the system, and V(x) represents the potential energy function that helps define the system. E is the energy of the system, and ψ is the function, called a wavefunction, that satisfies the differential equation.



David W. Ball
Schrödinger's equation is a second-order differential equation, and ψ is the function that, when substituted into the differential equation and evaluated, yields a constant (the energy E) times the original function. As you might suspect, not any random function will satisfy the second-order differential equation. In fact, for any given system and potential energy surface, there is only one family of functions that will satisfy the Schrödinger equation. These functions become the wavefunctions that define the behavior of the system.
Making matters worse, it is only for a few well-defined systems that a known, exact (so-called analytical) solution to the Schrödinger equation exists! For most systems, like atoms and molecules, the differential equation cannot be solved; that is, we don't know any mathematical formula that, when substituted into the Schrödinger equation, satisfies the differential equation.
Does that mean that the Schrödinger equation — and as a consequence, quantum mechanics — is useless? No, for two primary reasons. The first reason is that there are mathematical tools to approximate solutions to the Schrödinger equation to any level of accuracy we want. The second reason is that when we do these approximations, we still end up agreeing with experimental results. So quantum mechanics is still the most useful theory of the behavior of the atomic level to date.
Here, we will briefly review the ideal systems whose wavefunctions can be determined exactly using the Schrödinger equation. There won't be any long mathematical derivations; for most of these systems, adequate derivations and explanations can be found in any reasonable physical chemistry or quantum mechanics textbook.



Figure 1
The Particle in a One-Dimensional Box
Consider a particle moving between two infinitely high walls spaced by a width given by the variable a, as illustrated in Figure 1. The potential energy inside the box is zero. Because of this, the form of the Schrödinger equation that must be solved is


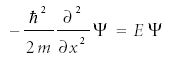
in the interval x = 0 to x = a. This is a relatively simple second-order differential equation, and functions that satisfy it are well known. They are
ψ = A sin Bx + C cos Dx
where A, B, C, and D are constants whose values will be dictated by the constraints on the system. Invoking the required traits of wavefunctions (they must be continuous, bound, single-valued, differentiable, normalized; see reference 3), we can determine that, to satisfy the constraints on the system, the only acceptable functions are


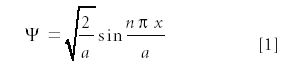
where a is the width of the box and n is a nonzero, nonnegative integer: 1, 2, 3, . . . A value of n = 0 is not allowed for mathematical reasons. The eigenvalue energy of the system can be determined by substituting ψ into the differential equation and solving:

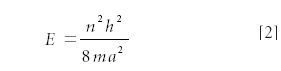
where all of the variables have been defined already. Because the integer n dictates the energy of the system, n is called a quantum number. Because the energy is based upon a collection of constants and an integral quantum number, we say that the energy is quantized.
Although the particle-in-a-box is an ideal system, it does have application in the real world. Some molecules have a long series of single and double carbon–carbon bonds called conjugated systems; the pi electrons in those double bonds act as if they extend the entire length of the conjugated system (Figure 2). Those electrons can be approximated as particles in a one-dimensional box, and their energy levels (as determined by spectroscopy) can be modeled using equation 2, shown previously.



Figure 2
The particle-in-a-box also can be extended to a three-dimensional system. Although it appears complex at first, the tactic is to treat each dimension separately, a technique called separation of variables. Each dimension then simplifies into the one-dimensional particle-in-a-box system, whose solution is shown earlier.


Figure 3
The Harmonic Oscillator
Consider a mass attached to a nonmoving wall by a spring (Figure 3). The mass can move back and forth under the influence of the spring, which works to return the mass to its equilibrium position during the course of the mass's motion. The force that the spring exerts on the mass is proportional to the displacement from the equilibrium position, and is always oriented opposite the mass's motion:
F = –kx
where x is the distance of the mass from the equilibrium position and k is called the force constant of the spring. The potential energy of the spring is given by the expression
V = ½ kx2
A repetitive motion whose force and potential energy follow these equations is called a harmonic oscillator. The harmonic oscillator was first described by Robert Hooke in 1660; the equations shown previously (especially the first one) are two forms of Hooke's law.
Quantum mechanically, a one-dimensional harmonic oscillator is treated with the Schrödinger equation, where in this case the potential energy is given by the expression mentioned earlier. The Schrödinger equation becomes


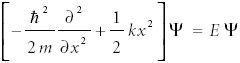
The presence of a nonzero potential energy term completely changes the differential equation.
The previous differential equation has a set of known solutions. They are


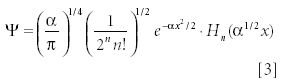
where


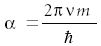
with ν itself being the classical frequency of the harmonic oscillator and defined as


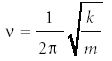
The "!" in the denominator of the second term implies the factorial function. The term Hn (α1/2 x) represents a family of polynomials called the Hermite polynomials. They are simply limited power series in x, having either all odd or all even powers of x in them. The index n represents the largest power of x in the polynomial. The first few are


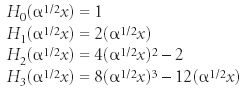
and so forth.
They were previously known functions that had application in probability theory; now we find them in quantum mechanics as well.
The wavefunction looks complex — but the energy eigenvalue is not. It is


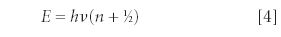
Note the presence of n, the index from the Hermite polynomial, in the expression. The energy of a quantum-mechanical harmonic oscillator is also quantized. One additional point to note — when n = 0, which is mathematically allowed in this system, the energy of the harmonic oscillator is not zero. Rather, it is ½hν, a quantity known as the zero point energy. The zero point energy is one of the more unusual predictions of quantum mechanics.
Atoms in compounds are connected with chemical bonds. These bonds also act like springs, and bonded atoms are constantly oscillating. These oscillations are treated as harmonic oscillators, and the harmonic oscillator system from quantum mechanics is applied to vibrations of atoms in molecules. Vibrational spectroscopy, which studies the vibrations of molecules, is a topic that has filled whole books. The vibrations of the water molecule, in which the centers of mass do not move but all of the atoms do, are shown in Figure 4.


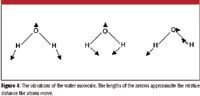
Figure 4
As useful as the harmonic oscillator is for studying vibrations, molecules are not perfect harmonic oscillators; they are anharmonic oscillators. There are several common ways of modifying the harmonic oscillator potential to accommodate a real, anharmonic oscillator. One of the most frequently seen anharmonic potentials is the Morse potential, which has the form
V(x) = De (1 – e–a(r – re))2
where De is the molecular dissociation energy and the constant a is defined as


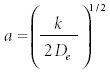
An anharmonic oscillator whose potential energy is described by the Morse potential also has a solvable Schrödinger equation. The eigenfunctions are


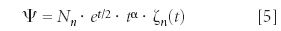
where Nn is a normalization constant, t = Ke–αx, K =


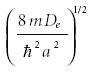
and ζn (t) is a polynomial function of t and K whose degrees depend on the quantum number n. The quantized energy eigenvalue is


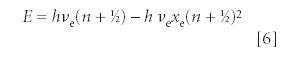
where νe is the classical harmonic frequency and xe is a dimensionless constant called the anharmonicity constant. Many references do not list xe, but νe xe instead. According to the solution of the Schrödinger equation, the anharmonicity constant xe equals (νe/4De).



Figure 5
Two-Dimensional Rotational Motion
Consider a particle of mass m constrained to motion in a circle having radius r (Figure 5). The potential energy on the particle is zero. This is now motion in two dimensions. In Cartesian coordinates, the Schrödinger equation is


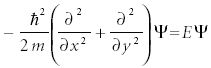
If we were to solve this version of the Schrödinger equation, we would have to solve for the two dimensions simultaneously, as a particle traveling in a circle is constantly changing its position in both dimensions. However, if we were to revise the coordinates of the system by defining an angle φ that the r vector makes with some arbitrary axis, the two-dimensional motion can be described more simply as the change of the angle φ. This coordinate system is called polar coordinates. The Schrödinger equation becomes


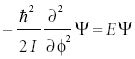
which is now a one-dimensional, second-order differential equation. The variable I is the moment of inertia, and for this simple system is equal to mr2.
This differential equation has known solutions — in fact, it is the same expression as the one we found in the particle-in-a-box system, only the boundary conditions are different. In this case, the eigenfunctions ψ are


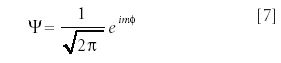
where i is the square root of –1 and the quantum number m is an integer — positive, negative, and zero values are allowed. The wavefunctions look like a sine wave curled around into a circle, with m positive and m negative extremes in the circle. The eigenvalue energies are quantized, and are given by


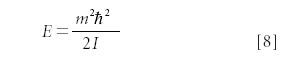
Note that the positive and negative values of |m| give the same value of energy, so for the first time we see an example of degeneracy, a situation in which two different quantum numbers yield the same energy. (The three-dimensional particle-in-a-box has degeneracies too, but we did not consider it there.) The positive and negative values of |m| correspond to the particles moving in opposite directions around the circle.
For the first time, another quantity is also quantized: angular momentum. The z-component of the angular momentum, Lz, is quantized and has values


Although an ideal system, two-dimensional rotation has applications to real systems. The pi electrons in benzene, C6H6, and other aromatic molecules can be thought of as moving about the molecule in a circle, and their energies can be approximated as two-dimensional rotational motion.


Figure 6
Three-Dimensional Rotational Motion
A particle of mass m moving on the surface of a sphere is rotating in three dimensions (Figure 6). If the potential energy is zero, then the Schrödinger equation to be solved is


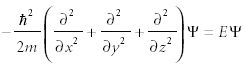
As with two-dimensional rotational motion, here we would have to solve for all three dimensions simultaneously. Also, as with two-dimensional motion, we transform the system into a coordinate frame based upon distance (r) and now, two angles: φ and θ. In these spherical polar coordinates, the Schrödinger equation is


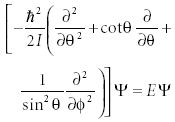
While this might be a more complicated form, it can be solved more easily than the Cartesian version by assuming a separable solution ψ = Θ(θ)·Φ( φ).
The solution to the Φ( φ) part is the same φ function that was the solution to the two-dimensional rotational motion


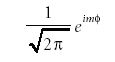
The solution to the Θ(θ) part is a set of polynomial functions called the associated Legendre polynomials. Like the Hermite polynomials, they are power functions, this time of sinθ and cosθ. Unlike the Hermite polynomials, the form of the Legendre polynomials is dictated by two indices, not one. The first index usually is labeled l, and because the second index has values that are limited by the first index, it is labeled ml. The relationship is
|ml| ≤ l
Thus, for any value of l, there are several (specifically, 2l + 1) possible values of ml. This is the same value of m that appears in the Φ part of the function as well. The first few associated Legendre polynomials (represented as Θ(l,ml)) are


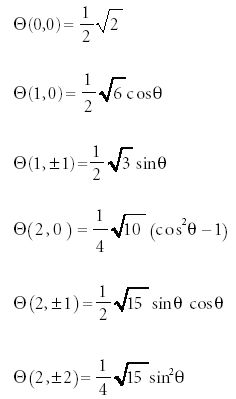
The products of the Φ and Θ functions in this system are called the spherical harmonics.
The expression for the quantized energy eigenvalue for three-dimensional rotation is

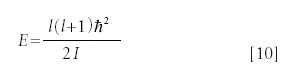
The square of the total angular momentum (not the total angular momentum itself) Ltot2, and the z component of the total angular momentum Lz also are quantized. Their values are


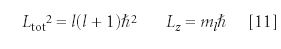
The electronic spectrum of buckminsterfullerene C60 can be approximated using three-dimensional rotational motion. With 60 pi electrons and an overall degeneracy of 2(2l + 1), all levels of l are filled through l = 4; the l = 5 level is the first partially filled level. The first electronic transition would be represented by l = 5 → l = 6; the calculated wavelength of 398 nm for the energy of this transition agrees well with an experimentally measured transition of 404 nm (4).
The rotations of molecules in the gas phase also are modeled using three-dimensional rotational motion. Agreement between theory and experiment actually is fairly good, and molecular parameters like bond angles and distance can be determined with some level of accuracy. Because molecules are not ideal systems, corrections for effects like centrifugal distortion sometimes are necessary to get theory and experiment to agree, especially for high-energy transitions.
The Hydrogen Atom
The movement from three-dimensional rotational motion to the hydrogen atom is a short one. There are only three major differences.
There are two particles, not one, in the hydrogen atom: the proton in the nucleus and the electron in orbit about the proton. Although the mass of the electron can be used directly, the reduced mass of the proton–electron system should be used instead (this only changes the result by about 0.05%).
The r value in the spherical polar coordinate system description of the H atom is not fixed.
The potential energy in the hydrogen atom is not zero. Rather, it is the coulombic potential energy that exists between two oppositely charged particles:


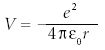
Thus, the complete Schrödinger equation for the hydrogen atom is


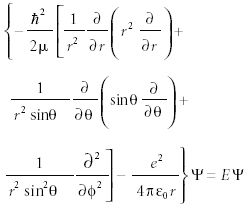
This differential equation is solvable if you assume a separable wavefunction, a product of a function of θ, a function of φ, and a function of r. It might be no surprise that the θ and φ functions are the spherical harmonics from three-dimensional rotational motion. The r function, also known as the radial function, is a series of functions known as the associated Laguerre polynomials. These polynomials are power functions of the variable r. The Laguerre polynomials introduce an index, n, that represents the highest power of r in the polynomial. This index also limits the possible values of l from the angular functions, which in turn limits the possible values of ml.
The eigenvalue energies of the hydrogen atom are quantized, and are given by the expression


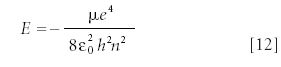
The variable μ is the reduced mass of the hydrogen atom, and ε0 is a constant called the permittivity of free space. The quantum number n comes from the Laguerre polynomial. Equation 12 is the same expression that Bohr derived in his theory of the hydrogen atom (see reference 2), except it did so under different assumptions, assumptions that are more defensible than those Bohr made. As such, quantum mechanics is a better theory.
Angular momenta also are quantized in the hydrogen atom just as they are in three-dimensional rotational motion, and they have the same possible values as given in equation 11. Rather than using a number for the quantum number l, a letter is used: s, p, d, f, . . . instead of 0, 1, 2, 3 . . .
Because it predicts the same expression for the energies of the atom that Bohr's analysis did, quantum mechanics also predicts the spectrum of the hydrogen atom — one of the fundamental failures of classical mechanics (see reference 1 for a discussion of the failures of classical mechanics). Equation 12 can be used to understand the energies of any one-electron system (He+, Li2+, and so forth) if allowances are made for the charge in the nucleus. Quantum mechanics gives us tools to understand other systems as well, something that Bohr's theory did not do.
Other Systems
After going through all of these systems and relating solutions to the various ideal systems, it might be hard to accept that there are no known analytic solutions of the Schrödinger equation to any systems with more than one electron. Even the helium atom, with only two electrons, does not have a solvable Schrödinger equation. The reason is that the Hamiltonian operator for the helium atom is not separable. The potential energy for helium includes a term for the electron–electron repulsion that occurs in the atom:


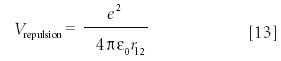
where r12 is the distance between the two electrons. The problem is that the value of r12 depends upon all three coordinates of each electron; that is, it is a six-dimensional variable. Thus, the problem cannot be separated into functions of each coordinate separately, so to date the differential equation that is the Schrödinger equation for the He atom has not been solved — nor will it ever likely be.
Does that mean that the helium atom's electrons do not have wavefunctions? No. Does that mean that quantum mechanics is useless? No again. Quantum mechanics does provide tools to understand systems larger than the hydrogen atom, and these tools are used the properties, behavior, and spectra of systems of any size.
The first tool is perturbation theory. Perturbation theory assumes that the Hamiltonian operator in the Schrödinger equation can be written as the sum of an ideal part, whose solutions are known, and an additive correction called the perturbation:


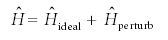
For example, in the helium atom, the ideal parts can be hydrogen-like electrons and the perturbation can be the interelectron repulsion given in equation 13. Then, the average energy can be determined (see Part III for a discussion of average values of observables):


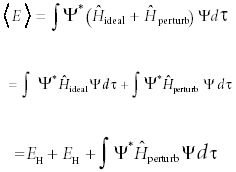
The first two terms are simply the energies of two hydrogen electrons. The last term is the additive perturbation, which can always be evaluated either analytically or numerically (after all, an integral is simply the area under a curve). In the case of helium, a total energy is calculated that is within about 5% of the experimental value.
Corrections to wavefunctions also can be defined. Details can be found in any decent textbook that covers quantum mechanics. The point, however, is that it can be done using perturbation theory.
The second tool is variation theory. This is based upon the variation theorem, which essentially states that any appropriate trial wavefunction for a system will always yield an average energy that is equal to or greater than the actual energy. As such, we can take a trial function and give it one or more constants whose value is unknown. We then determine an expression for the average energy, and then determine values for the constants that yield the lowest value for the energy. Those values of the constants give us the best possible wavefunction of that formula.
Again, let us use helium as an example. Although the helium atom has a nucleus that has a 2+ charge, it is doubtful that each electron experiences the full 2+ charge because of the presence of the other electron; this concept is known as shielding. Let us then use a hydrogen atom wavefunction as a trial function, but let the nuclear charge Z vary and see what value of the nuclear charge gives us the lowest energy for the helium atom. A moderate amount of calculus demonstrates that the lowest energy occurs with a nuclear charge Z = 27/16, which is slightly less than 2. Using this method, we can determine an energy that is within 2% of the experimental value of helium.
Table I summarizes our calculated values of the energy of helium. Granted, there is still some error between theory and experiment, but remember: classical mechanics could not even predict this level of accuracy!



Table I: Calculated and experimental values of the He atom, using the tools of quantum mechanics
Using perturbation theory and variation theory in conjunction with experiments, scientists and spectroscopists study the energy levels of atoms and molecules. By defining various perturbations and including multiple variables within functions, scientists can model an atomic or molecular system to any accuracy desired. Other issues apply: symmetry, selection rules, the Born–Oppenheimer approximation, and other issues that could be topics of future columns. But they all fall back on quantum mechanics. So the next time you measure a spectrum, think of yourself as a "quantum mechanic" because spectroscopy really is applied quantum mechanics.
David W. Ball is a professor of chemistry at Cleveland State University in Ohio. Many of his "Baseline" columns have been reprinted in book form by SPIE Press as The Basics of Spectroscopy, available through the SPIE Web Bookstore at www.spie.org. His most recent book, Field Guide to Spectroscopy (published in May 2006), is available from SPIE Press. He can be reached at d.ball@csuohio.edu his website is academic.csuohio.edu/ball.
References
(1) D.W. Ball, Spectroscopy 22(12), 91–94 (2007).
(2) D.W. Ball, Spectroscopy 23(1), 18–22 (2008).
(3) D.W. Ball, Spectroscopy 23(4), 14–17 (2008).
(4) D.W. Ball, J. Chem. Educ. 71, 463 (1994).
















LIBS Illuminates the Hidden Health Risks of Indoor Welding and Soldering
April 23rd 2025A new dual-spectroscopy approach reveals real-time pollution threats in indoor workspaces. Chinese researchers have pioneered the use of laser-induced breakdown spectroscopy (LIBS) and aerosol mass spectrometry to uncover and monitor harmful heavy metal and dust emissions from soldering and welding in real-time. These complementary tools offer a fast, accurate means to evaluate air quality threats in industrial and indoor environments—where people spend most of their time.


NIR Spectroscopy Explored as Sustainable Approach to Detecting Bovine Mastitis
April 23rd 2025A new study published in Applied Food Research demonstrates that near-infrared spectroscopy (NIRS) can effectively detect subclinical bovine mastitis in milk, offering a fast, non-invasive method to guide targeted antibiotic treatment and support sustainable dairy practices.

Smarter Sensors, Cleaner Earth Using AI and IoT for Pollution Monitoring
April 22nd 2025A global research team has detailed how smart sensors, artificial intelligence (AI), machine learning, and Internet of Things (IoT) technologies are transforming the detection and management of environmental pollutants. Their comprehensive review highlights how spectroscopy and sensor networks are now key tools in real-time pollution tracking.


