The Tiger Has Sharp New Teeth
The new FDA Commissioner wants a strong FDA and is backing her words with action by initiating a program that cuts the time that firms must respond to 483 observations from 30 to 15 business days. Not only is the time halved but the response must be complete. Therefore it is better and cheaper to be compliant than not.



The Food and Drug Administration (FDA) has not had good press over the last few years, as they have been on the receiving end of two critical reports from the Government Audit Office (GAO). The last report covered the FDA foreign inspection program and was damning in the fact that the FDA does not know how many establishments it has to inspect, relies upon volunteers to inspect overseas, and when they do conduct inspections, the frequency is lower than in the U.S. (1).



R.D. McDowall
FDA Modernization Act 2009
As a result of this report and also the increased threat of supply chain contamination, the FDA Modernization Act 2009 (2) is being passed into law and covers the following areas:
- Creates an up-to-date registry of all drug and device facilities serving American consumers.
- Increases funding for more GMP inspections for ethical and generic drugs as well as introduces preapproval inspections for generic drugs.
- Requires parity between foreign and domestic inspections, and to meet this, the FDA is setting up offices in China, India, Europe, and Latin America.
- Denies entry to drugs coming from facilities that limit, delay, or deny FDA inspections.
- Requires manufacturers to know their supply chain including the identification and mitigation of risk throughout their supply chain.
- Requires country-of-origin labeling for components.
Concomitantly, there also has been an increase in budget to fund this and increase the inspection program. So what does that mean for me in the laboratory? This stuff appears to be too high a level to even contemplate doing anything about. What's this about a tiger and new teeth? It may seem that the tiger has been stuffed but the good bits start in the next section.
New FDA Commissioner Acts
In addition to the FDA Modernization Act 2009, there also has been a change at the top with the appointment of Dr. Margaret Hamburg as the new FDA commissioner in May 2009. On August 6, 2009, the new commissioner made a speech that emphasized the need for a "strong FDA," which highlighted the benefits of this new approach as having credibility with the public, being transparent in explaining its decisions, being able to enforce the law, and being creative in promoting health. In this speech, with an accompanying video (3), were such quotations as
- "Through regular inspections and follow-up on signals indicating problems, the FDA must work to identify and resolve problems early."
- "Companies must have a realistic expectation that if they are crossing the line, they will be caught, and that if they fail to act, we will."
- "The agency must show industry and consumers that we are on the job. We must publicize our enforcement actions — and the rationale for those actions — widely and effectively."
- "The agency must place greater emphasis on significant risks and violations, and use meaningful penalties to send a strong message to discourage future offenses."
The strong message coming through loud and clear is be compliant and remain compliant with the regulations — or else. You'll now see that the tiger, far from being ready for the taxidermist, is starting to sharpen its teeth.
Not only does this apply to the here-and-now but also, as noted in the final bullet point, the commissioner wants to discourage future offences. So why change the FDA approach to enforcement? One of the rationales for this was mentioned in the speech as the pathways for enforcement action can be too long and arduous when the public's health is in jeopardy. So from this and the FDA, we must infer that the FDA will be increasing inspections while tightening up and speeding up enforcement actions.
Thus. it was not surprising that the FDA announced in the Federal Register of August 11 (4) the trial of a new program entitled "Review of Post-Inspection Responses." The purpose of this program is to facilitate the timely issuance of warning letters that started on September 15, 2009 and will run for 18 months (the tiger has just come back from the dentist and is feeling hungry — are you getting the message?).
Issues with the Current Inspection Process
Before we look in detail at the inspection goodies coming your way, I would just like to step back and review what happened before implementation of this new program. FDA inspections come in three flavors and will continue to do so:
- Facility inspection: routine inspection of the facility and the six areas quality — however, we will just focus on the quality system and the laboratory in this column.
- Preapproval inspection (PAI): inspection of the manufacturing facility for a new drug or a generic drug before licensing.
- For cause inspection: either there is a public health issue that must be rapidly investigated or a whistleblower has alerted the agency. In either case, the first thing you'll probably notice is the knock of the inspection team at the front door of your site.
Regardless of the inspection type, the inspection follows the same format and begins with the inspectors presenting their credentials and a copy of FDA Form 482, which is the notice of inspection, at the opening meeting. During the inspection, they may notice noncompliances and these are documented on FDA Form 483 entitled "Inspectional Observations" (this is the dreaded 483!), which is given to the company at the closing meeting of the inspection. Written on each page of Form 483 is the following wording:
"This document lists observations made by the FDA representative during the inspection of your facility. They are inspectional observations, and do not represent a final agency determination regarding your compliance."
The company currently has 30 working days to respond to the 483 but what happens in practice is that a first response is presented within the time limit and then further letters are fed in over a few months for the more serious issues. If you read some of the warning letters on the FDA website, you'll see if the inspection occurred in January, the warning letter finally emerges into the light 6–9 months later. The reason for this is that there has been protracted correspondence between the company and the FDA. This will stop under the new program.
Warning letters are only issued for significant violations from the regulations. After the warning letter has been sent out to the company, a copy is hung on the "Wall of Shame" in the warning letter section of the FDA's website. Here anyone can have a good laugh about the problems of another company while secretly hoping that they will not have a starring role in a future warning letter. The response time for the company to respond to a warning letter is 15 working days; in the new program, this also will be the response time for a 483.
Regardless of whether you get a 483 or not, the FDA inspectors will write a detailed Establishment Inspection Report (EIR) of the inspection that details the people they had talks with and the procedures, systems, and records that were inspected; this document provides the agency with its elephant's memory. Failure to correct the noncompliances outlined in a warning letter may lead to further enforcement action by the FDA, and ignoring a warning letter is not advisable. However, the agency has often issued further warning letters to companies in the past, a practice that will apparently stop with the new program. Further regulatory enforcement by the FDA can include a consent decree of permanent injunction, which will bind the company in perpetuity to comply with the regulations; this can cost a company millions of dollars or even its existence or withdrawing a product licence. For foreign companies, the FDA can ban import of its products.
483 Complaints Procedure
Under the "GMP for the 21st Century" initiative, the FDA issued a guidance for industry with a delightful title of "Formal Dispute Resolution: Scientific and Technical Issues Related to Pharmaceutical CGMP" (5). In essence, this document encourages the company and the inspector to discuss and resolve everything while the inspection is still ongoing. However, if this is not possible, only with regard to observations where it is down to interpretation is a company allowed to appeal to the FDA in a formal, two-tiered response. For example, if you don't keep complete records of a spectroscopic analysis, you will not have a leg to stand on as the regulations explicitly require you to do this. Here you are noncompliant and you have been caught — no discussion and no argument. In contrast, if the regulations are vague and it is down to differences of interpretation between the company and the inspector, then this is where the dispute procedure can be used.


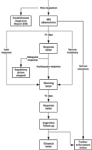
Figure 1: Process flow of the new FDA postinspection response program.
Postinspection Response Program
So what has changed with this new program? Two changes that will impact laboratories is that a very short timeframe is being put around how quickly firms must respond to 483 observations, and the second is that there will be a formal close out of warning letters to demonstrate that the noncompliances listed there have been resolved. So let's look in detail at what you have to do and you will now see that the tiger has sharp new teeth and that they will be sinking into your flesh. Figure 1 shows the process as a diagram and readers should look at this to put the text below into context.
- FDA has now set postinspection deadlines for responses by organizations. The response time for firms to respond has been halved to 15 working days. This is not just to send a letter to begin a cozy correspondence with the agency but to have a complete response to all the 483 observations. If the compliance problems are significant, the timeframe for the response is critical and the response must be full, adequate, and complete. The letter will not be able to show that the problems have been resolved for all but the simplest observations but will demonstrate how they will be resolved and in what timeframe. If a response is not received in this time or the response is inadequate, then the agency can start work on a warning letter or other enforcement action. We'll take a look at ways to respond to a 483 or warning letter in a later section of this column.
An important point to understand here is that the FDA now takes the systems-based approach to inspections and coupled with ICH Q10 on pharmaceutical quality systems (6) you will need a different approach to respond to 483 observations. If there is an observation that has an impact outside of the area inspected, you will be expected to fix it as part of the systems-based approach. Remember, the wording at the end of virtually every warning letter on the FDA website: "this letter is not intended to be an all-inclusive list of the violations at your facility. It is your responsibility to ensure compliance with applicable laws and regulations administered by FDA." However, there is an onus on the inspector to discuss findings with management to minimize surprises and errors when issuing the 483 at the inspection closing meeting.
- The warning letter issuing process is being streamlined and speeded up by the FDA. Instead of the FDA lawyers reviewing all warning letters, only those where there are significant legal issues will now get the legal eagle eye. The agency will not delay issuing a warning letter if you are late in responding to the 483. Even if your response to the 483 is perfect, the FDA does "not plan to routinely include a response on the apparent adequacy of the firm's corrective actions in the warning letter (4)." Instead, the agency will respond to evaluate both your response to the 483 and the warning letter together. Therefore, the message is clear to all: Your 483 response must be timely and complete, or else you could end up with a warning letter on the front doormat pretty quickly afterwards.
- Next, the FDA will prioritize enforcement follow-up. After a warning letter is issued or a major product recall occurs, the FDA will make it a priority to follow up promptly with appropriate action, such as an inspection or investigation to assess whether or not a company has made required changes in its compliance practices.
- In the past, it was not uncommon for a company to have a number of warning letters after several inspections; it may be the companies involved thought that they could manage the warning letters, the FDA, and still do business. However, under this new program, the agency will not issue multiple warning letters like confetti to noncompliant firms before taking greater enforcement action. The agency will move quickly and aggressively to address significant health concerns or serious violations by immediate action — even before they have issued a formal warning letter. This message is crystal clear — be compliant or there will be regulatory trouble and the FDA will move to protect public health.
- In a novel approach to noncompliance, the FDA will now issue a close-out letter to companies issued warning letters to document that they have made necessary corrections. As a copy of this close-out will be posted on the FDA website alongside the warning letter, readers will be able to see how quickly companies respond to the regulator. It is hoped that this will be an important motivator for companies to fix the problem quickly.
So does the FDA have sharp new teeth or are they toothless? It is your call to decide, but as a word of advice, be compliant and inspection-ready in your laboratory.
Are You Inspection-Ready?
Remember, the cost of compliance is always, always, cheaper than the cost of noncompliance. You can decide how to do things rather than rush through something with a regulator breathing your neck. Therefore, the easiest way to respond to a 483 observation is to be compliant and don't get one. However, we have to manage risk in the industry and this is probably unrealistic. So, don't get a serious 483 observation. You'll need to have a quality system that is defendable, as this is always better than arguing with the inspector on the day of the inspection. Rather than just sit there and wait for the next inspector to drop, all staff should be vigilant and ensure that they work in a compliant manner and — this point is very important — improve the current processes and the associated compliance. This is not just a job for quality assurance personnel who conduct internal audits and document checks, it is everybody's job. This internal effort must be coupled with an increased program of laboratory audits aimed at verifying what the real level of compliance is in your organization. It is all well and good knowing that in six months time an inspection is due and that you can prepare for it. When the inspector comes around the laboratory is clean and tidy and all the staff are in their Sunday best. However, what is the reality of the day-to-day operations?
Here, audits can be a very effective means to determining the real level of compliance. Not just planned audits that analytical scientists know will occur and can prepare for but unannounced audits that give the real level of compliance and inspection readiness of a laboratory. This latter type of audit can be disruptive but is a vital means of avoiding the 15-day post-inspection panic that will become a feature of noncompliant organizations in the coming months. Also, from personal audit experience, it is very useful to use a digital camera to record some types of noncompliance such as untidy laboratories or missing calibration stickers on equipment as the auditee has less wiggle room when the evidence is in the report. To be effective, audits need to be conducted by trained staff external to the laboratory under audit to remove bias from the results.
Responding Effectively to a 483 Observation
Ok, so you ignored the advice above and now you have one or more 483 laboratory observations — so what are you going to do? Remember, the clock starts ticking 15 days from the closing meeting so if you want to avoid the FDA tiger taking a bite out of various portions of your anatomy, you had better start thinking and quickly. A good option is to talk through the observation with the inspector while he or she is still on site to clarify the issue and to inform the inspector what action will be taken to resolve the problem. This should help you understand the problem and to get feedback if this is a way to bring you back into compliance.
The response to the agency must be formal and that is typically with a covering letter written at a minimum by the site head or better still by the chief executive officer. This is intended to demonstrate to the FDA how seriously the firm takes the situation and that it also provides a written commitment to comply voluntarily.
This will be coupled with the attachments discussing how the company will resolve the inspection observations. The documents may be sent initially by fax or e-mail but it always should be followed up with a hard copy couriered to the field office. To write the attachments, you will need to gather the experts in the organization who will help draft the response to specific observations. Also, you should know when you have to obtain external expertise to help you. This is especially important with the new program, as you only have a maximum of 15 days before the time limit is up and external resources may need time to mobilize. The attachments will detail how you will resolve each of the observations or noncompliances. It is extremely important that these are laid out clearly: therefore, address each observation at the beginning of a new page. Ensure also that the response numbering is the same as the observation numbering in the 483. This makes it easier for the official at the FDA to correlate the material (remember your reader!).
For each observation:
- Do you agree or disagree with the inspector's observation?
Acknowledging that this is correct means you understand the noncompliance. Coupled with this needs to be an understanding of the underlying regulation that has caused the observation: Is your interpretation correct and how has this been documented in your SOPs?
- A root cause analysis may be important for some observations as the underlying cause of the observation may not be immediately apparent in many cases.
- Either provide:
A description of a completed corrective action with evidence of the work carried out.
A corrective action plan with evidence of work carried out to date.
A corrective action plan with sufficient detail and a reasonable timescale outlining what will be done and by when. Will you be working with external experts who will help and guide you in your corrective action plan?
- Make sure that the work package to resolve the observation is specific and complete: for example, if an SOP is to be updated as there is an omission, provide a copy of the new SOP but also outline what training was provided to the impacted staff with copies of the material and updated training records.
- Realism is important as the FDA officials reviewing your response are not idiots. As you cannot validate a laboratory information management system (LIMS) in one month, don't expect the FDA to believe you if you put this in your response letter. Credibility is a vital commodity here — do not lose it. Therefore, be able to ensure that you can deliver on the plan and have the resourcing to do this. Strangely, at times like this, words such as "budget" and "constraint" are never found together in the same sentence.
- Addressing just the observation is like putting a band-aid on a broken leg: you need to understand what the impact is on other parts of the organization, other products, methods, instruments, systems, and so forth. This is the systems-based approach. For larger organizations, it may not be at just one site but globally. Remember, it is the EIR that will provide the FDA with ammunition to look at other sites and if they find the same problems — you are in trouble.
- It's all well and good providing plans and schedules for doing the work. How do you know the work will resolve the problem? More importantly, how do you know if the problem will not reoccur in the future? Therefore, you must anticipate these questions from the agency: How will you verify that the work has been done, what documentation will be available to demonstrate this, and how will you monitor the area to ensure that it does not happen again?
A well-constructed response letter delivered within the new 15-day timeframe will go a long way toward restoring your regulatory credibility. Still, you could look on the bright side: The program is an 18-month trial, so will the FDA drop this approach after the trial ends? I don't think so, look at the resume of the new commissioner, read the speech and watch the video. I think this new program is here to stay. We will see an initial increase in warning letters but these will tail off as industry ensures it stays in compliance but it will be a rough time initially, as the tiger builds up momentum with its sharp new teeth.
Summary
In this column, I have looked at the new postinspection response program, which was introduced on an 18-month trial by the FDA on the September 15, 2009. The tiger's new teeth are sharp and you had better beware — complete responses to the 483 observations are required by 15 working days after the inspection or a warning letter or increased enforcement action may ensue. I have also outlined a practical way for audits to check you are in compliance but if that fails, a means to respond to 483 observations. But remember, time is not on your side here.
R.D. McDowall is principal of McDowall Consulting and director of R.D. McDowall Limited, and "Questions of Quality" column editor for LCGC Europe, Spectroscopy's sister magazine. Address correspondence to him at 73 Murray Avenue, Bromley, Kent, BR1 3DJ, UK.
References
(1) "Better Data Management and More Inspections are Needed to Strengthen FDA's Foreign Drug Inspection Program," U.S. Government Accountability Office, Report GAO-08-970, September 2008.
(2) HR 759 FDA Modernization Act 2009.
(3) "Law Enforcement and Benefits to Public Health," Margaret Hamburg, 6th August 2009. A copy of the speech is available at http://www.fda.gov/NewsEvents/Speeches/ucm175983.htm and the video of the speech can be accessed on the same page.
(4) Review of Post-Inspection Responses Federal Register, August 11, 2009, 74 (153) 40211–40212.
(5) "Guidance for Industry, Formal Dispute Resolution: Scientific and Technical Issues Related to Pharmaceutical CGMP," FDA January 2006.
(6) International Conference on Harmonisation (ICH) Q10: Pharmaceutical Quality Systems step 4 (see www.ich.org).















