Atomic and Molecular Emissions Observed from Mid-Infrared Laser-Induced Breakdown Spectroscopy
The authors use a novel MIR laser-induced breakdown spectroscopy (LIBS) probe, biochemicals, and inorganic alkali metal salts to produce emissions in the MIR region from atomic and oxygenated molecular breakdown species.



By using a novel mid-infrared (MIR) laser-induced breakdown spectroscopy (LIBS) probe, biochemicals and inorganic alkali metal salts produced emissions in the MIR (2–5.75 μm) region from atomic and oxygenated molecular breakdown species. MIR LIBS CO and CO2 emissions resulted from oxidation of laser-sputtered carbon fragments from carbon-containing poly(3-hydroxybutyrate) and sodium carbonate. Moreover, intense IR emissions were detected from atomic transitions between high-lying Rydberg states of laser-induced breakdown products from inorganic sodium carbonate alkali metal salt tablets. The MIR emitting species was determined to be the neutral alkali atoms.
Laser-induced breakdown spectroscopy (LIBS) recently has emerged as a powerful spectroscopic tool for chemical analysis and identification of trace elements because of its simplicity and versatility. In LIBS, a high peak energy density of focused, intense laser pulses is used to ionize and atomize analyte molecules via multiphoton excitation and cascade bremsstrahlung (1). This results in a hot and short-lived (on the order of 100 ns) plasma created at the analyte material surface. The induced plasma is characterized by high electron and ion densities, and temperatures typically reach 105 K. As the laser-induced plasma quickly expands outward and cools, the electrons and ions recombine, and a characteristic loud sound can be heard due to the shock wave emitted from the plasma. Valuable information on the identification and concentration of the trace materials can be acquired by monitoring atomic and molecular emissions from the consequential relaxation processes of the excited atomic, ionic, and molecular fragments in the plasma. There are several major advantages of LIBS: no time-consuming sample preparation needed; the ability to perform real-time, on-site analysis because the laser-induced plasma prepares and excites the sample in a single step; and the potential for remote sensing (2) and field-portable sensors (3). Its ability to analyze samples rapidly and directly without cumbersome sample preparation has led to its promising applications in environmental science (4), space missions (5), medicine (6), industry (7), and homeland security (8).
Nearly all the analytical LIBS applications reported in the literature so far have only exploited the emissions in the UV–vis–near IR (NIR) region (emission wavelengths shorter than 2 μm) from atoms and very simple molecular fragments such as OH and C2. However, it is well known that molecules reveal spectroscopic signatures in the longer wavelength MIR region due to molecular vibrational and rotational transitions. MIR emission spectra of materials have greater complexity and richness compared with the UV–vis region. The relatively more complex MIR emission spectra have the potential to provide greater detection and differentiation characteristics. Therefore, an extension of LIBS to the MIR region promises to provide additional information concerning the identification and classification of analyte substances, which can augment conventional UV–NIR LIBS measurements. Furthermore, the MIR LIBS analysis is a more logistically convenient in-situ probe than the long-established infrared emission spectroscopic techniques such as flame spectroscopy and pyrolysis. Another advantage of MIR LIBS with respect to the conventional IR emission techniques is its capability and simplicity for time-resolved investigation of breakdown reaction products. Being a multiphoton–bremsstrahlung process, it is also more tolerant to pump laser wavelength selection than laser-induced infrared fluorescence spectroscopy.
Several studies have suggested the existence of more complicated molecular breakdown fragments generated from laser-induced plasma. In LIBS studies of various organic compounds, UV–vis emission features from C2 were found to be correlated strongly to the presence of aromatic rings in the analyte material (9) and aromatic ring arrangements (10). Studies using laser desorption mass spectrometry also have shown the laser-induced vaporization (ablation) process to provide adequate molecular fragments for identification of a variety of chemical and biological species (11). Conventional flame-induced breakdown emission spectroscopy applications produce molecules that are excited, atomized, and ionized by combustion thermal energy transfer. Emission features from at least three different types of molecular breakdown products including oxygenated fragments, hydrogenated fragments, and partially disintegrated molecule fragments can be observed and monitored readily in the molecular vibrational and rotational MIR region (12). Those studies illustrate the potential for recovering MIR molecular emission features from a LIBS event.
In our recent attempts to explore the feasibility of using MIR spectroscopy to probe the emissions of molecular products from reactions initiated by laser-induced plasmas, MIR atomic (13) and molecular (14) emissions from solid analyte were identified for the first time. The MIR (between 3 and 5.75 μm) LIBS molecular emissions of the laser-induced breakdown products, CO and CO2 molecules from solid organic substrates, have been observed (14). The hot CO2 and CO molecules originate from the oxidation of laser-sputtered carbon species from the organic analyte. Moreover, intense infrared emissions between 2 and 5.6 μm were detected from atomic transitions between high-lying Rydberg states of laser-induced breakdown products from various alkali metal halide tablets (13). The MIR emitting species was determined to be the neutral alkali atoms from the metal halide. These high-intensity, narrow linewidth MIR atomic emissions should be valuable in the interpretation of the overall LIBS spectra in the MIR region.
The work presented here intends to expand upon those previous MIR LIBS studies by probing organic biochemicals and solid inorganic targets (Na2CO3) that contain both alkali metals and carbon oxides between 2 and 5.75 μm.
Experimental
The MIR LIBS experimental setup has been reported elsewhere (14). The 1064-nm output of a Q-switched Nd:YAG laser (Surelite II, Continuum Electro-Optics, Inc., Santa Clara, California) with a pulse width of 5–10 ns and a 10-Hz repetition rate was used as an excitation source for the MIR LIBS setup. The pulsed laser beam was focused onto the sample surface using a plano-convex lens (f = 20 cm), resulting in a beam size diameter less than 0.2 mm. A reflective beamsplitter (~4%) was employed to monitor the laser beam energy during the emission measurements. The laser energy throughout the experiment was found to be very stable, with less than 1% fluctuation. The laser energy reaching the sample surface was about 16 mJ/pulse and was sufficient to produce plasma for all compounds tested, which included paper cellulose fiber (white index card), glass (microscope slide), and poly(3-hydroxybutyrate) and sodium carbonate tablets made from high-pressure application of the vacuum-dried powders.
Figure 1 presents a diagram of the experimental setup. The samples were mounted vertically onto a linear translation stage with a stepping motor (Aerotech, Inc., Pittsburgh, Pennsylvania) and translated at a speed of ~1.5 mm/min during each emission scan so that the laser pulse would always hit a fresh spot. The speed of the spectral emission scan rate was set to 100 nm/min from 2000 nm to 5750 nm. The MIR LIBS emission was collimated and focused onto the entrance slit of a 0.15-m grating spectrometer (λblaze = 4 μm, 150 g/mm) (Princeton Instruments-Acton, Trenton, New Jersey) using two CaF2 focusing lenses (f = 15 cm, f = 20 cm). The entrance and exit slits of the spectrometer were adjusted to ~1 mm for the biochemical studies and 0.25 nm for the inorganic salts. The reason for using a smaller slit width for the salt studies was to have a higher resolution of the intense emissions of the MIR atomic lines. A 2-μm long-pass filter was placed in front of the entrance slit to prevent laser scattering from entering the spectrometer. The spectral resolution was approximately 40 nm and 10 nm for the biochemical studies and inorganic salt, respectively. The signal was recorded and analyzed using a liquid nitrogen-cooled InSb (Judson Technologies, LLC, Montgomeryville, Pennsylvania) detector and a temporal gated boxcar averager (Stanford Research Systems, Sunnyvale, California). Spectra typically were acquired at time delays between 10 μs and 40 μs after the laser pulse, and the gate width was fixed at 16 μs.


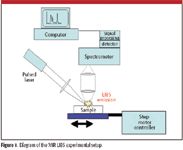
Figure 1
The MIR emission spectra were corrected for the spectral response of the experimental setup and atmospheric gas absorptions by using a calibrated blackbody radiation source (Optronic Laboratories, Inc., Orlando, Florida). The spectrum of the calibration lamp was measured under identical experimental parameters (slit width, long pass filter) as employed during the MIR LIBS studies. The wavelength accuracy of the experimental setup was determined using the well-known CO2 absorption features centered at 4.25 μm (2350 cm–1).
Results and Discussion
MIR LIBS Spectra of Biochemicals
The MIR emission transients of organic compounds were found in our previous studies (13,14) to be characterized by a short-lived initial spike (0–5 μs) followed by a slower decaying component (0–300 μs). The temporal behavior of the initial fast spike was limited by the detector response time (~ 5 μs). A spectrum of the initial spike was clearly dominated by the hot plasma continuous blackbody emission due to electron–ion recombination and collisions between electron and excited atomic and molecular fragments. The peak maximum of the slower decaying emission component occurred at 50 μs. Emission captured in the time period between 40 and 56 μs after the initial plasma formation clearly revealed no trace of the blackbody continuum and a strong molecular emission feature between 3 nm and 5.75 nm that was attributed to CO2 and CO vibrational emissions.
In this work, we extend the spectral range of our recently developed MIR LIBS probe (14) to 2–5.75 μm and apply it on an intracellular polymer, poly(3-hydroxybutyrate), that is produced by certain microorganisms. Figure 2 shows the MIR LIBS emission spectra of the poly(3-hydroxybutyrate), paper cellulose fiber, and glass slide silicate between 2 and 5.75 μm captured between a 40 and 56 μs time delay. The spectra show no trace of thermal background emission, and the spectra of the carbon-containing samples show emission features between 2400 nm and 3000 nm and 4200 nm and 4800 nm. There was no comparable strong emission feature from the noncarbon, glass slide. The strong emission features spanning from 4200 to 4800 nm from both organic compounds have been identified elsewhere (14). These emission bands could be attributed to an overlap of the CO stretching vibration around 4700 nm (15) and CO2 asymmetric stretching vibration around 4360 nm (16) resulting from the oxidation of laser-sputtered carbon fragments from the organic solid samples.



Figure 2
In the region between 2400 nm and 3000 nm, emission features between 2.5 and 2.8 μm from poly(3-hydroxybutyrate) and at 2.5, 2.75, and 2.9 μm from the paper fiber are observed. The 2.5-μm feature in the paper fiber spectrum is broader and weaker than that in the poly(3-hydroxybutyrate) spectrum. In the flame spectroscopy studies of methanol and oxyacetylene, combustion products such as H2O emit vibrational bands at 2, 2.48, and 2.7 μm; CO emits at 2.3 μm; and CO2 emits at 2.8 μm (12,17). Therefore, we can attribute the 2.5-μm emissions of both compounds to the R branch of the OH-stretching vibrations of molecular water species. The emission features between 2.7 and 3 μm could be from the 2.78- and 2.9-μm vibrational overtones of CO2 and the Q and P branch OH-stretching vibrations (2.8 μm) of molecular water species. The hot emitting H2O species should originate from the oxidation of laser-sputtered hydrogen fragments from the organic solid samples. It appears that the emission contribution from ambient atmospheric CO2 and H2O is minimal to negligible, because the silicate spectra in Figure 2 yielded no CO2 or H2O emission peaks. There are other candidates that could contribute to the emission in this region, such as amide A bands. Additional study is required to further resolve the origin of the 2.7–3 μm bands. We also noticed that the H2O emission bands at 2 and 5.2 μm, present in the flame spectra of methanol and oxyacetylene, are below the detection limits of the present experimental setup.
MIR LIBS Spectra of Sodium Carbonate
Recently, the observation of LIBS MIR atomic emission lines between 2 and 5.6 μm from various neutral alkali metal laser breakdown fragments from solid alkali metal halide tablets has been reported (13). In this work, we use MIR LIBS to study a carbon-containing alkali salt for the presence of both atomic and molecular vibrational emissions in the MIR region.



Table I: The assignments of MIR LIBS emission features between 2 and 5.75 μm from sodium carbonate
We observed five distinct MIR LIBS emission features from sodium carbonate. The assignments of the MIR LIBS emission features from sodium carbonate are listed in Table I. The distinct emission features at 2.1, 2.3, 3.4, and 4.0 μm originated from the neutral sodium atom 42P3/2, 42D3/2, 42D5/2, and 52G7/2,9/2 energy levels (13). The broader 4.4-μm bands of sodium carbonate can be assigned to asymmetric stretching vibrations of CO2 molecules (14). Figure 3 illustrates the temporal emission curves of the 2.21-μm peak (solid) and 4.4-μm peak (dashed) from solid sodium carbonate. Both transient emission spectra were characterized by a short-lived initial spike (0–5 μs blackbody continuum) followed by a slower decaying component. These are observed as a 15-μs shoulder for the 2.21-μm peak (solid curve in Figure 3) and a broad feature centered at 40 μs for the 4.4-μm peak (dashed curve in Figure 3).


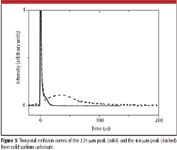
Figure 3
Figure 4 provides MIR spectra of the sodium carbonate sample at various time delays after plasma formation. The laser input energy was 16 mJ, and a 16-μs gate width was applied. Note that from 10 to 40 μs delay, the relative magnitudes of the atomic emission signatures decrease and those of the molecular spectral signatures increase.



Figure 4
During the first few microseconds, the 1064-nm laser pulse initiates white-hot plasma and vaporizes and atomizes the sodium carbonate molecules and atomic (sodium and carbon) breakdown species mainly by cascade electron–atom collisions (1). In the next 10 μs, while the plasma expands and cools, the excited atomic species relax from a series of atomic transitions between higher-lying Rydberg states and emit signature light in the MIR region. In the meantime, some hot atomic fragment (such as carbon) might collide and react with ambient atmospheric oxygen, and for the next 100 μs, these reactions will continue and produce oxygenated molecular breakdown species such as CO2. Those hot molecular breakdown species then emit vibrational signatures in the MIR region until they cool to equilibrium after a few hundred microseconds.
Summary
By using a novel MIR LIBS probe, we investigated a biochemical and an inorganic alkali metal salt and successfully identified emissions from atomic and oxygenated molecular breakdown species in the MIR (2–5.75 μm) region. MIR emission spectroscopy was used to probe molecular products from reactions initiated by laser-induced plasmas. The observation of these MIR atomic lines together with the vibrational and rotational emission features from the laser breakdown of solid compounds demonstrated the promise of MIR LIBS as a potent complement to conventional UV–NIR LIBS as a powerful chemical analysis probe. Although emission signatures from partially dissociated molecular fragments were not detected, the wavelength region between 2 and 5.75 μm is the so-called quiet region for infrared signature absorption and emission. More spectral regions, especially between 8 and 12 μm, will be mined for the appearance of MIR LIBS emissions that might be directly indicative of partially dissociated and recombination molecular species.
Clayton S.-C. Yang is with Battelle Eastern Science and Technology Center, Aberdeen, Maryland. Ei Brown and Uwe Hommerich are with the Department of Physics, Hampton University, Hampton, Virginia. Sudhir B. Trivedi is with Brimrose Corporation of America, Baltimore, Maryland. Alan C. Samuels and A. Peter Snyder are with Edgewood Chemical Biological Center, Aberdeen Proving Ground, Maryland.
References
(1) F.-Y. Yueh, J.P. Singh, and H. Zhang, "Laser-Induced Breakdown Spectroscopy, Elemental Analysis" in Encyclopedia of Analytical Chemistry: Applications, Theory, and Instrumentation. R.A. Meyers, Ed. (John Wiley & Sons Ltd, Chichester, 2000), pp. 2066–2088.
(2) K. Stelmaszczyk, P. Rohwetter, G. Méjean, J. Yu, E. Salmon, J. Kasparian, R. Ackermann, J.-P. Wolf, and L. Wöste, Appl. Phys. Lett. 85, 3977–3979 (2004).
(3) R.S. Harmon, F.C. DeLucia, C.E. McManus, N.J. McMillan, R. Coveney, T.F. Jenkins, M.E. Walsh, and A. Miziolek, Appl. Geochem. 21, 730–747 (2006).
(4) K.Y. Yamamoto, D.A. Cremers, M.J. Ferris, and L.E. Foster, Appl. Spectrosc. 50, 222–233 (1996).
(5) R. Brennetot, J.L. Lacour, E. Vors, A. Rivoallan, D. Vailhen, and S. Maurice, Appl. Spectrosc. 57, 744–752 (2003).
(6) O. Samek, M. Liska, J. Kaiser, D.C.S. Beddows, H.H. Telle, and S. Kukhlevsky, J. Clin. Laser Med. Surg. 18, 281–289 (2000).
(7) A.S.G. Buckley, H.A. Johnson, K.R. Hencken, and D.W. Hahn, Waste Management 20, 455–462 (2000).
(8) A.C. Samuels, F.C. DeLucia Jr., K.L. McNesby, and A.W. Miziolek, Appl. Opt. 42, 6205–6209 (2003).
(9) L. St-Onge, R. Sing, S. Bechard, and M. Sabsabi, Appl. Phys. A 69, 913–916 (1999).
(10) A. Portnov, S. Rosenwaks, and I. Bar, Appl. Opt. 42, 2835–2842 (2003).
(11) R.C. Beavis and B.T. Chait, Rapid Commun. Mass Spectrom. 3, 432–435 (1989).
(12) C.K.Y. Lam, D.C. Tilotta, K.W. Busch, and M.A. Busch, Appl. Spectrosc. 44, 318–325 (1990).
(13) C.S.-C. Yang, E.E. Brown, U.H. Hommerich, S.B. Trivedi, A.C. Samuels, and A.P. Snyder, submitted to Appl. Spectrosc.
(14) C.S.-C. Yang, E.E. Brown, U.H. Hommerich, S.B. Trivedi, A.C. Samuels, and A.P. Snyder, Appl. Spectrosc. 61, 321–326 (2007).
(15) M.K. Hudson and K.W. Busch, Anal. Chem. 60, 2110–2115 (1988).
(16) D.C. Tilotta, K.W. Busch, and M.A. Busch, Appl. Spectrosc. 43, 704–709 (1989).
(17) Flame Spectroscopy, R. Mavrodineanu and H. Boiteux, Eds. (John Wiley and Sons, New York, 1965).














AI Shakes Up Spectroscopy as New Tools Reveal the Secret Life of Molecules
April 14th 2025A leading-edge review led by researchers at Oak Ridge National Laboratory and MIT explores how artificial intelligence is revolutionizing the study of molecular vibrations and phonon dynamics. From infrared and Raman spectroscopy to neutron and X-ray scattering, AI is transforming how scientists interpret vibrational spectra and predict material behaviors.


Real-Time Battery Health Tracking Using Fiber-Optic Sensors
April 9th 2025A new study by researchers from Palo Alto Research Center (PARC, a Xerox Company) and LG Chem Power presents a novel method for real-time battery monitoring using embedded fiber-optic sensors. This approach enhances state-of-charge (SOC) and state-of-health (SOH) estimations, potentially improving the efficiency and lifespan of lithium-ion batteries in electric vehicles (xEVs).


New Study Provides Insights into Chiral Smectic Phases
March 31st 2025Researchers from the Institute of Nuclear Physics Polish Academy of Sciences have unveiled new insights into the molecular arrangement of the 7HH6 compound’s smectic phases using X-ray diffraction (XRD) and infrared (IR) spectroscopy.

