Consequences of Finite Ion Lifetimes in Mass Spectrometry
Consequences of Finite Ion Lifetimes in Mass Spectrometry As we construct more-complex instruments that process packets of ions in time and space, the issue of ion lifetimes is becoming more important.



An advertising slogan suggests that "diamonds are forever," leading to some interesting discussions of long-term thermodynamic stability in freshman chemistry classes. For mass spectrometry (MS), the finite lifetime of organic ions has practical consequences. Simply stated, we want ions, once formed, to survive unchanged until we either force a change in mass, charge, or structure (as in MS-MS analysis), or until the ions arrive at the instrument detector after mass analysis. Instead of millions of years for the lifetime of diamonds, our focus is shifted to a lifetime scale usually ranging from microseconds to milliseconds for ions derived from organic and biochemical compounds. As we construct more-complex instruments that process packets of ions in time and space, the issue of ion lifetimes grows in importance. On one end of the scale, groups of specialists use MS to measure the masses of very short-lived radioactive nuclides. On the other end of the scale, our ability to store atomic ions for very long periods in ion traps leads to fundamental experimental determinations in metrology.
In teaching organic mass spectrometry (MS) with electron ionization, we usually invoke the Franck–Condon principle (1–3, for various spectroscopic applications) to describe the timing perspective of the ionization process and subsequent passage of the ions through the mass spectrometer. Classically, the Franck–Condon principle states that the electronic transition that leads to ionization of the molecule occurs much faster than any changes in the bonding or positions of the atoms of the molecule. The molecular ion thus (at least initially) retains the structure of the molecule from which it is formed. Ions are accelerated "promptly" from the source into the mass analyzer. If we completed the story of the timing framework, me might suggest that ions pass so quickly through the instrument that the relatively slow rate at which we scan a mass analyzer (as in a sector-based mass spectrometer) is of no practical consequence.
We use the kinetic energy equation to calculate the velocity of ions moving through the MS system as an example of this framework. In any mass spectrometer, the accelerating potential of the source V provides the ion with a potential energy that is equal to the kinetic energy, defined as (1/2)mv2 , where m is the mass of the ion in kilograms and v is the velocity in meters per second. The calculational exercise (Table I) is especially useful in that it links ion masses in terms of daltons (Da) to ion masses in kilograms and also provides a real sense of the speed of ions as they pass through a laboratory-scale instrument. Table I collects calculated results for a variety of ions, with a range of masses and charges, and their calculated velocities and passage through a 1-m linear flight path in a time-of-flight (TOF) instrument with 5000 V accelerating potential. The calculation easily leads to a discussion of where an ion spends most of its time in a mirror TOF system and to further consideration of the length of ion flight paths in other mass analyzers.


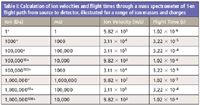
Table I: Calculation of ion velocities and flight times through a mass spectrometer of 1-m flight path from source to detector, illustrated for a range of ion masses and charges
As MS instruments for organic analyses were developed, scientists scanning the baseline observed reproducible signals of different characters than the normal stable ions in the mass spectrum. The signals were metastable ions (4). A metastable ion appears in the mass spectrum obtained with a magnetic-sector mass analyzer as a low-intensity diffuse peak (Figure 1), contrasted with the sharp peaks for stable ions drawn from the source of the mass spectrometer and dispersed in normal fashion by the magnetic sector. A metastable signal arises due to the dissociation of a parent ion to a product ion, after acceleration from the source, and in a field-free region before entrance into the mass analyzer. This physical region is termed a field-free region because it is not subject to the acceleration potential that draws ions from the source and the ion has not yet entered the mass analyzer field. It is not strictly field-free, as focusing elements may be placed there, but the term persists. In a Nier-Johnson geometry instrument, parent ions m1 that dissociate in the first field-free region to a product ion m2 constitute a metastable ion m* observed in the mass spectrum at an apparent mass of m* = (m22 /m1). Furthermore, the kinetic energy release associated with the dissociation shapes the peak so that the observed signal is broad compared to "normal" peaks. Figure 1 shows metastable ions observed between ions of m/z 14 and m/z 26 in such an instrument. The sharp peaks (representing ion beams for "normal" fragment ions) contrasts sharply with the diffuse peak shapes of the metastable ion signals. Metastable ions are a fingerprint of fragmentation reactions, just as those that lead to the "normal" mass spectrum. The insightful early works into their origins and use were notable. These studies led to determinations of the half-lives of ions and measurements of the few percent that dissociate in metastable transitions (5), to the inference of molecular ions when not otherwise observed (6), and to stereochemical determinations for ions (7).


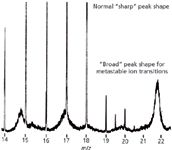
Figure 1: Different peak shapes observed for normal ions (sharp) and metastable ion transitions (broad) within the low-mass range on a Nier-Johnson instrument. (Created from Figure 15 in reference 4.)
Instruments for Studying Metastable Ions
Although many of these early studies of metastable ions derived from organic compounds were carried out with sector-based instruments, the ions don't "know" about the nature of the instrument used to study them. Metastable ions dissociate according to their structure, charge state, and internal energy, and our instruments may or may not be able to observe these reactions. Let's consider metastable ions in a TOF instrument. In the idealized TOF source, all ions are formed in the same place at the same time and are accelerated through the same potential into the flight tube. Their race through the flight tube is timed, with the lighter mass ions arriving at the detector first (Table I). What happens if an ion dissociates after acceleration into the flight tube but before arrival at the detector? The fragment ions and neutral species continue moving through the flight tube at essentially the same velocity as the dissociating parent ion (8). The signal generated by the detector is therefore due to the coincident arrival of a combination of undissociated parent ions, fragment ions, and even fast neutral species. Standing discusses the significance of the ion-to-neutral conversion in his personal review of TOF-MS (9). Szymczak and Wittmaack discuss the process of ion-to-neutral conversion in secondary ion MS with a TOF instrument (10). Neutral-ion coincidence measurements can increase the signal-to-noise ratios in the MS-MS spectrum recorded with a TOF instrument (11).
The early study of metastable ions and the later redevelopment of TOF instruments for biomolecular analysis at higher masses helped to reveal the diversity of dissociation reactions of energized ions as they move through mass spectrometers — through a certain physical space (the flight path) or within a certain period in a constrained trap (the flight time). While we implicitly expect the ions to be stable, the ions may have other ideas. The advent of MS instruments other than sector-based instruments, and the almost universal use of computerized data acquisition, may diminish our recognition that metastable ion dissociations are inevitable. Although the footprint of MS instruments has been reduced, the total flight path and flight time for ions can be surprisingly long, allowing ample opportunity for ion dissociation. For example, a quadrupole mass filter may be approximately 25 cm in physical length, but ions follow an oscillating orbital path through the filter that is longer. Also, in the quadrupole mass filter, the ions are not accelerated to as high a velocity as in a sector-based instrument and are moving more slowly. In an ion-trap or a Fourier-transform (FT)-MS instrument, the ions orbit in the center of the trap and may remain there for milliseconds, or much longer in specialized experiments. In an Orbitrap instrument (Thermo Fisher Scientific, Waltham, Massachusetts), the ions are kept in the trap until the desired mass measurement accuracy is achieved, sometimes for seconds. In a TOF instrument with a mirror, the flight path is lengthened because the ions travel from the source to the mirror and then from the mirror to the detector. In any instrument, the velocities of the ions may change slightly with focusing elements, or much more drastically with deceleration–acceleration schemes. For instance, in a mirror TOF instrument, the ions spend a seemingly disproportionate time within the mirror; as the ion velocities are slowed their directional vector changes and the ions are then reaccelerated.
Hybrid Instruments
In the quest for higher performance, manufacturers now offer a variety of "hybrid" instruments for MS-MS. Hybrid mass spectrometers are instruments constructed with at least two component "mass" analyzers of different types, arranged in sequence from ion source to ion detector. Hybrid mass spectrometers can be composed of beam components (an ion beam moves through the component) or trap components (a packet of ions resides within the component). In hybrid instruments, the concepts of flight path and flight time become even more complex and the ions have ample opportunity to dissociate when we do not expect them to. Time (as in determining a flight time) is a malleable parameter in a hybrid mass spectrometer because ions can be transferred between hybrid instrument components in discrete packages and independent processes can occur in independent components. In a trap–trap hybrid instrument, for example, the final trap can be processing a population of product ions by mass selection or ion activation, or proving an accurate mass measurement, while the initial trap can be processing single-stage mass spectral data, or even completing lower resolution MS-MS analyses in its own right. Even as we construct such instruments for discrete and separate stages of ion activation and analysis, a certain percentage of the ions will dissociate on their own at some point. Because the instrumental parameters are set to pass or preserve stable ions, these metastable ions are lost to further processing, invariably reducing the overall instrument sensitivity.
Is there anything that can be done to reduce the metastable ion loss? As discussed in a previous column (12), pressure and vacuum are not really trivial. Seemingly small changes in pressure lead to large changes in ion transmission through instrument components simply as a result of scattering. Factor in processes such as collision-induced dissociation and the consequential effect grows. In a complex hybrid instrument, careful attention to the pressure profile along the ion flight path is crucial. Better pumping, leading to a lower pressure at crucial locations in the instrument, is almost always guaranteed to lead to better overall performance. Recent strategies for sampling directly at atmospheric pressure generate their own set of engineering challenges for the pumping system of the mass spectrometer. Although it may be possible to operate an instrument at "higher than optimal" pressures engendered by the atmospheric pressure sampling, its performance will invariably be degraded.
In contrast to ions derived from organic and biological compounds, atomic ions have no paths to dissociation and more limited paths to neutralization. Long-lived atomic ions have been measured in mass spectrometers to determine their accurate masses. For example, the high-mass-measurement accuracy of the FT-MS instrument, used to great advantage in organic and biomolecular analysis, translates to atomic ion mass measurement as well (13). Introduction and maintenance of single ions within a Penning trap instrument, and mass measurements with a high degree of precision and accuracy, is a fundamental metrological experiment (14). Such experiments inform the basic foundations on which measurement science is based (15), but are unfortunately not broadly recognized within the community devoted increasingly to biomolecular analysis. However, the fundamental measurement of mass for short-lived radioactive nuclides depends explicitly on MS (16), and the exacting procedures developed in such work to minimize errors are instructive for all researchers in the field.
Conclusion
Finally, we note that trapped ions can be evaluated not for their mass, but as an optical clock (17). The mass of a single trapped ion is a constant, whether it is measured or not. An atomic clock has been devised based on optical transitions between trapped 199 Hg+ and 27 Al+ ions. This high-accuracy clock represents a new basis for the measurement of time itself. We close this column with a quote (17): "The uncertainties in the atomic clocks reported here occur at the exciting intersection of relativity, geodesy, and quantum physics, and the total uncertainty of 5.2 × 10-17 shows unprecedented sensitivity to gravitational effects and cosmological fluctuations. Future improvements in these atomic clocks will provide even more sensitive probes of nature." Cosmological fluctuations? I always thought that instrument performance was somehow tied to phases of the moon, and I may have been right all along.
Kenneth L. Busch is amused to think that, at one extreme, our experiments define time with such precision and accuracy that we can watch for cosmological fluctuations. On another level, Samoa is scheduled to leap back across the International Dateline at the end of this year, losing an entire day (and a Friday at that!). Their last change was in 1892, apparently accomplished without too great an inconvenience. In 2011, we assume there's already an ID hop app, and if there's not, sooner or later (pun intended), there will be. This column is the sole responsibility of the author, who can be reached at WyvernAssoc@yahoo.com.


Kenneth L. Busch
References
(1) S.E. Schwartz, J. Chem. Educ. 50, 608–610 (1973).
(2) R.L. Dunbrack, Jr., J. Chem. Educ. 63, 953–955 (1986).
(3) F. Ahmed, J. Chem. Educ. 64, 427–428 (1987).
(4) R.G. Cooks, J.H. Beynon, R.M. Caprioli, and G.R. Lester in Metastable Ions (Elsevier, Amsterdam, 1973). (This book has been reprinted by the American Society for Mass Spectrometry.)
(5) J.A. Hipple, Phys. Rev. 71, 594–599 (1947).
(6) L.A. Shadoff, Anal. Chem. 39, 1902–1903 (1967).
(7) K.C. Kim and R.G. Cooks, J. Org. Chem. 40, 511–513 (1975).
(8) C.R. Ponciano and E.F. da Silveira, J. Phys. Chem. A 106, 10139–10143 (2002).
(9) K.G. Standing, Int. J. Mass Spectrom. 200, 597–610 (2000).
(10) W. Szymczak and K. Wittmaack, Appl. Surf. Sci. 203–204, 170–174 (2003).
(11) F.H. Strobel and D.H. Russell, Anal. Chem. 64, 2879–2881 (1992).
(12) K.L. Busch, Spectroscopy 24(11), 16–22, (2009).
(13) M.V. Gorshkov, S.H. Guan, and A.G. Marshall, Int. J. Mass Spectrom. Ion Phys. 128, 47–60 (1993).
(14) R.C. Thompson, Meas. Sci. Technol. 1, 93–105 (1990).
(15) F. DiFillipo, V. Natarajan, K.R. Boyce, and D.E. Pritchard, Phys. Rev. Letters 73(11), 1481–1484 (1994).
(16) http://isoltrap.web.cern.ch/isoltrap/
(17) T. Rosenband, D.B. Hume, P.O. Schmidt, C.W. Chou, A. Brusch, L. Lorini, W.H. Oskay, R.E. Drullinger, T.M. Fortier, J.E. Stalnaker, S.A. Diddams, W.C. Swann, N.R. Newbury, W.M. Itano, D.J. Wineland, and J.C. Bergquist, Science 319, 1808–1812 (2008).














High-Speed Laser MS for Precise, Prep-Free Environmental Particle Tracking
April 21st 2025Scientists at Oak Ridge National Laboratory have demonstrated that a fast, laser-based mass spectrometry method—LA-ICP-TOF-MS—can accurately detect and identify airborne environmental particles, including toxic metal particles like ruthenium, without the need for complex sample preparation. The work offers a breakthrough in rapid, high-resolution analysis of environmental pollutants.


The Fundamental Role of Advanced Hyphenated Techniques in Lithium-Ion Battery Research
December 4th 2024Spectroscopy spoke with Uwe Karst, a full professor at the University of Münster in the Institute of Inorganic and Analytical Chemistry, to discuss his research on hyphenated analytical techniques in battery research.

Mass Spectrometry for Forensic Analysis: An Interview with Glen Jackson
November 27th 2024As part of “The Future of Forensic Analysis” content series, Spectroscopy sat down with Glen P. Jackson of West Virginia University to talk about the historical development of mass spectrometry in forensic analysis.

Detecting Cancer Biomarkers in Canines: An Interview with Landulfo Silveira Jr.
November 5th 2024Spectroscopy sat down with Landulfo Silveira Jr. of Universidade Anhembi Morumbi-UAM and Center for Innovation, Technology and Education-CITÉ (São Paulo, Brazil) to talk about his team’s latest research using Raman spectroscopy to detect biomarkers of cancer in canine sera.


