Linearity in Calibration: Quantifying Nonlinearity, Part II
At this point in our series dealing with linearity, we have determined that the data under investigation do indeed show a statistically significant amount of nonlinearity, and we have developed a way of characterizing that nonlinearity. Our task now is to come up with a way to quantify the amount of nonlinearity, independent of the scale of either variable, and even independent of the data itself.



In our last few columns (1–5), we devised a test for the amount of nonlinearity present in a set of comparative data (for example, as are created by any of the standard methods of calibration for spectroscopic analysis), and then pointed out a flaw in the method. The concept of a measure of nonlinearity that is independent of the units that the X and Y data have is a good one. The flaw is that the nonlinearity measurement depends upon the distribution of the data; uniformly distributed data will provide one value, normally distributed data will provide a different value, randomly distributed (i.e., what is found commonly in "real" data sets will give still a different value, and so forth, even if the underlying relationship between the pairs of values is the same in all cases.
"Real" data, in fact, might not follow any particular describable distribution at all. Or the data might not be sufficient to determine what distribution it does follow, if any. But does that matter? At the point we have reached in our discussion, we already have determined that the data under investigation do indeed show a statistically significant amount of nonlinearity, and we have developed a method of characterizing that nonlinearity in terms of the coefficients of the linear and quadratic contributions to the functional form that describes the relationship between the X and Y values.
Our task now is to come up with a way to quantify the amount of nonlinearity the data exhibits, independent of the scale (that is, units) of either variable, and even independent of the data itself. The first condition is met by converting the nonlinear component of the data to a dimensionless number (that is, a statistic), akin to but different than the correlation coefficient, as shown in our previous column (5).
The second condition can be met simply by ignoring the data itself, once we have reached this point. What we need is a standard way to express the data so that when the statistic in computed, the standard data expression will give rise to a given value of the statistic, regardless of the nature of the original data.
For this purpose, then, it would suffice to replace the original data with a set of synthetic data with the necessary properties. What are those properties? The key properties comprise the number of data values, the range of the data values, and their distribution.
The range of the synthetic data we want to generate should be such that the X-values have the same range as the original data. The reason for this is obvious — when we apply the empirically derived quadratic function (found from the regression) to the data, to compute the Y-values, those should fall on the same line, and in the same relationship to the X as did the original data.
Choosing the distribution is a little more nebulous. However, a uniform distribution is not only easy to compute, but also will not go outside the specified range, nor will the range change with the number of samples, as data following other distributions might (see for example reference 6 or chapter 6 in reference 7, where we discussed the relationship between the range and the standard deviation for the Normal distribution when the number of data differ, although our discussion was in a different context). Therefore, in the interest of having the range and the nonlinearity measure be independent of the number of readings, we should generate data following a uniform distribution.
The number of data points to generate in order to get an optimum value for the statistic is not obvious. Intuition indicates that the value of the statistic might very well be a function of the number of data points used in it's calculation. At first glance, this also would seem to be a "showstopper" in the use of this statistic for the purpose of quantifying nonlinearity. However, intuition also indicates that even so, the use of "sufficiently many" data points will give a stable value, Because "sufficiently many" eventually becomes an approximation to "infinity," and therefore even in such a case will at least tend toward an asymptotic value, as more and more data points are used. Because we already have extracted the necessary information from the actual data itself, computations from this point onward are simply a computer exercise, needing no further input from the original data set.
Therefore, in fact, the number of points to generate is a consideration that in itself needs to be investigated. We do so by generating data with controlled amounts of nonlinearity as we have previously (5), and by filling in the range of the X-values with varying numbers of data points (uniformly spaced), computing the corresponding Y-values (according to the computed values for the coefficients of the quadratic equation), and then the statistic we described previously (5). We performed this computation for several different combinations of number of data points generated and the value of k, the nonlinearity term generator from Equation 1 in the previous column. The results are shown in Table I. Although not shown, similar computations were performed for 200,000 and 1,000,000 points. There was no further change in any of the entries, compared to the column corresponding to 100,000 points.



Table 1.
As we can see, the value of the nonlinearity value converges to a limit for each value of k, as the number of points used to calculate it increases. Furthermore, it converges more slowly when the amount of nonlinearity in the data increases. The results in Table I are presented to four figures, and to require that degree of convergence means that fully 10,000 points must be generated if the value of k approaches 2 (or more). Of course, if k is much above 2, it might require even more points to achieve this degree of exactness in the convergence. For k = 0.1, however, this same degree of convergence is achieved with only 500 points.
Thus, the user must make a trade-off between the amount of computation performed and the exactness of the calculated nonlinearity measure, taking into account the actual amount of nonlinearity in the data. However, if sufficient points are used, the results are stable and depend only upon the amount of nonlinearity in the original data set.
Or need the user do anything of the sort? In fact, our computer exercise is just an advanced form of a procedure that we all learned to do in second-term calculus — evaluate a definite integral by successively better approximations, the improvement coming via exactly the route we took — using smaller and smaller intervals at which to perform the numerical integration. By computing the value of a definite integral, we are essentially taking the computation to the limit of an in finite number of data points.
Generating the definite integral to evaluate is in fact a relatively simple exercise at this point, since the underlying functions are algebraic. We recall that the pertinent quantities are
- The sum of squares of the differences between the linear and quadratic lines fit to the data; and
- The sum of squares of the Y-data linearly related to the X-data.
As we recall from the previous column (5), the nonlinearity measure we devised equals the first divided by the second. Let us now develop the formula for this. We will use a subscripted small "a" for the coefficients of the quadratic equation, and a subscripted small "k" for those of the linear equation. Thus, the equation describing the quadratic function fitted to the data is


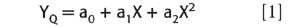
The equation describing the linear function fitted to the data is



Where the ai and the ki are values obtained by the least-squares fitting of the quadratic and linear fitting functions, respectively. The differences, then, are represented by


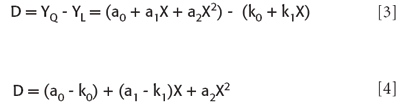
and the squares of the differences are


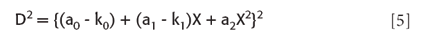
which expands to


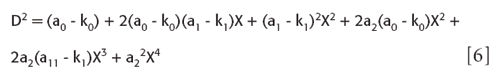
we can simplify it slightly to a regular polynomial in X:


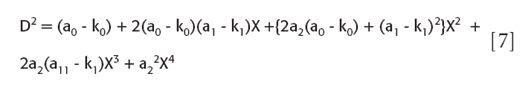
The denominator term of the required ratio is the square of the linear Y term, according to equation 4. The square involved is then



and substituting for each Y the expression for X:


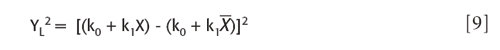
With a little algebra this can also be put into the form of a regular polynomial in X:


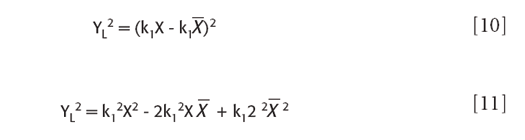
which, unsurprisingly, equals:



although we will find equation 11 more convenient. Equations 7 and 11 represent the quantities whose sums form the required measurement. They are each a simple polynomial in X, whose definite integral is to be evaluated between Xlow and Xhigh, the ends of the range of the data values, in order to calculate the respective sums-of-squares.
Despite the apparently complicated form of the coefficients of the various powers of X in equation 7, once they have been determined as described in our previous column, they are constants. Therefore, the various coefficients of the powers of X are also constants, and may be replaced by a new label, we can use subscripted small "c" for these; then equation 7 becomes


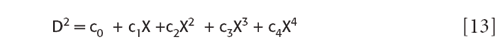
Put into this form it is clear that forming the definite integral of this function (to form the sum of squares) is relatively straightforward, we merely need to apply the formula for the integral of a power of a variable to each term in equation 13. We recall that from elementary calculus the integral of a power of a variable is:



Applying this formula to equation 13, we achieve:


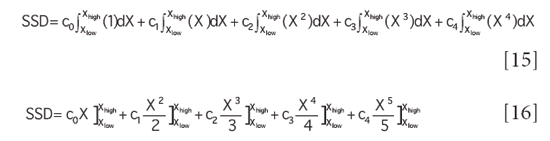
where the various ci represent the calculation based on the corresponding coefficients of the quadratic and linear fitting functions, as indicated in equation 7.
The denominator term for the ratio is derived from equation 11 in similar fashion; the result is


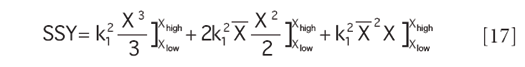
and the measure of nonlinearity, then, is the result of equation 16 divided by equation 17.
Why Linearity Is Important
It will be helpful at this point to again review the background of why nonlinearity is important to understand why we bring up the "News Flash." In the context of multivariate spectroscopic calibration, for many years most of the attention was on the issues of noise effects (noise and error in both the X (spectral) and Y (constituent values) variables. The only attention paid to the relation between them was the effect of the calibration algorithm used, and how it affected and responded to the noise content of the data. There is another key relationship between the X and Y data, and that is the question of whether the relationship is linear, but that is not addressed. In fact, few people talk (or write) about it even though it is probably the only remaining major effect that influences the behavior of calibration models. A thorough understanding of it probably would allow us to solve most of the remaining problems in creating calibrations. A nonlinear relation potentially can cause larger errors than any random phenomenon affecting a data set (see, for example, reference 8). The question of linearity inevitably interacts with the distribution of constituent values in the samples (not only of the analyte but of the interferences as well, see reference 8). Our attention first was turned to this issue back when multiple linear regression (MLR) was king of the near infrared (NIR) hill, and we couldn't understand how the wavelength selection process worked, and why it picked certain wavelengths that appeared to have no special character. The Y-error was the same for all sets of wavelengths. The X-error might vary somewhat from wavelength to wavelength, but the precision of the NIR instruments was so good that the maximum differences in random absorbance error simply couldn't account for the variations in the wavelengths chosen. Eventually, the realization arose that the only explanation that was never investigated was that a wavelength selection algorithm would find those wavelengths where the fit (in terms of linearity of the absorbance versus constituent concentrations) of the data could change far more than any possible change in random error. Considerations of nonlinearity potentially explains lots of things: the inability to extrapolate models, the "unknown" differences between instruments that prevents calibration transfer, and so forth. Recently, we wrote some columns that showed that principle component regression (PCR) and partial least squares (PLS) also are subject to the effects of nonlinearity and are not simply correctable (9–14). So there is a big effect here that few are paying attention to — at least not insofar as they are quantitatively evaluating the effect on calibration models. This is key, because it is inevitably one of the major causes of error in the X-variable (at least, as long as the X-variable represents instrument readings).
Now here's the news flash: we recently became aware that Philip Brown has written a paper (15) nominally dealing with wavelength selection methods for calibration using the MLR algorithm (more about this paper later). We're old MLR advocates (since 1976, when we first got involved with NIR and MLR was the only calibration algorithm used in the NIR world then). But what has happened is that until fairly recently, the role of nonlinearity in the selection of wavelengths for MLR, as well as other effects on the modeling process has been mostly ignored (and only partly because MLR itself has been ignored until fairly recently). For a long time, however, there was much confusion in the NIR world over the question of why computerized wavelength searches would often select wavelengths on the side of absorption bands instead of at the peaks (or in other unexpected places), and manual selection of wavelengths at absorption peaks would produce models that did not perform as well as when the wavelengths on the side of the peaks were used. This difference existed in calibration, validation, and in long-term usage. It also was (and still is, for that matter) independent of the methods of wavelength selection used. This behavior puzzled the NIR community for a long time, especially because it was well-known that a wavelength on the side of an absorbance band would be far more sensitive to small changes in the actual wavelength measured by an instrument (due to nonrepeatability of the wavelength selection portion of the instrument) than a wavelength at or near the peak, and we expected that random error from that source should dominate the wavelength selection process. In hindsight, of course, we recognize that if a nonlinear effect exists in the data, it will affect the modeling process implicitly, regardless of whether the nonlinearity is recognized or not.
The Mysteries of NIR
There are other "mysteries" in NIR (and other applications of chemometrics) that nonlinearity also can explain. For example, as indicated earlier, one is the difficulty of transferring calibration models between instruments, even of the same type. Where would our technological world be if a manufacturer of, say, rulers, could not transfer the calibration of the unit of length reliably from one ruler to the next?
But here's what Philip Brown did: He took a different tack on the question. He set up and performed an experiment wherein he took different sugars (fructose, glucose, and sucrose) and made up solutions by dissolving them in water, each at five different concentration levels, and made solutions using all combinations of concentrations. That gave an experimental design with 125 samples. He then measured the spectra of all of those samples. Because the samples were all clear solutions there were no extraneous effects due to optical scatter.
The nifty thing he then did was this: he then applied an analysis of variance (ANOVA) to the data, to determine which wavelengths were minimally affected by nonlinearity. We have discussed ANOVA in this column also (16–19), although, to be sure, our discussions were at a fairly elementary level. The experiment that Philip Brown performed is eminently suitable for that type of computation. The experiment was formally a three-factor multilevel full-factorial design. Any nonlinearity in the data will show up in the analysis as what statisticians call an "interaction" term, which can even be tested for statistical significance. He then used the wavelengths of maximum linearity to perform calibrations for the various sugars. We'll discuss the results below, because they are at the heart of what makes this paper important.
This paper by Brown is very welcome. The use of this experimental design is a good way to analyze the various effects he investigates, but unfortunately is not applicable to the majority of sample types that are of interest in "real" applications, where neither experimental designs nor nonscattering samples are available or can be generated. In fact, it can be argued that the success of NIR as an analytical method largely is due to the fact that it can be applied to all those situations of interest in which neither of those characteristics exist (in addition to the reasons usually given about it being nondestructive). Nevertheless, we must recognize that in trying to uncover new information about a technique, "walking before we run" is necessary and desirable, and this paper should be taken in that spirit.
Especially because Brown explicitly considers and directly attacks the question of nonlinearity, which is a favorite topic of ours (in case you couldn't tell), largely because it's mostly been ignored previously as a contributor to the error in calibration modeling, and because the effects occur in very subtle ways — which is largely what has hidden this phenomenon from our view.
Overall it's a wonderful paper, despite the fact that there are some criticisms. The fact that it directly attacks the issue of nonlinearity in NIR is one reason to be so pleased to see it, but the other main reason is that it uses well-known and well-proven statistical methodology to do so. It is delightful to see classical statistical tools used as the primary tool for analyzing this data set.
Because we tend to be rather disagreeable sorts, let's start by disagreeing with a couple of statements Brown makes. First, while discussing the low percentage of variance in the 1900-nm region accounted for by the sugars, he states, "... where there is most variability in the spectrum and might wrongly be favored region in some methods of analysis." We have to disagree with his decision that using the 1900-nm region is "wrong." This is a value judgment and not supported by any evidence. To the contrary, he is erroneously treating the water component of the mixtures as though it had no absorbance, despite his recognition that water, and the 1900-nm region in particular, has the strongest absorbance of any component in his samples. Why say this? Because of the result of combining two facts:
- The system is closed in that the total concentrations of all four components add to 100%, and also because the total variance due to all four components (and interactions) add to 100%.
- The water not only has absorbance, it is the strongest absorber in the mixtures.
If water had no absorbance — that is, if it was the "perfect nonabsorbing solvent" that we like to deal with — then Brown's statement would be correct. It would not contribute to the variance and the three sugars would be the source of all variance. But in that case, the total variance in the 1900-nm region would also be less than it actually is, so we cannot say a priori what would happen "if." But we can say the following: because the absorbance of the water in that wavelength region is strong, we can consider the possibility that a measurement there will be a (inverse, to be sure) measure of "total sugar" or some equivalent. However, the way the experiment is set up precludes a determination of the presence of nonlinearity of the water absorbance in that region. If it were linear, then it should be determinable with the least error of all four components, because it has the strongest absorbance, and therefore, any fixed amount of random error would have the least relative effect. Then it would be a matter of determining which two sugars could be determined most accurately, and then the third by difference. This essentially is what he does for the linear effects he analyzes, so this would not be breaking any new ground, just using the components that are most accurately determined to compute all concentrations. But to get back to where this all came from, this is the reason we disagree with his statement that using the water absorbance is "wrong."


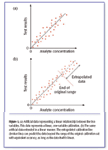
Figure 1.
Now let us do a thought experiment, as illustrated in Figure 1 (reproduced from reference 5): imagine a univariate spectroscopic calibration (with some random error superimposed) that follows what essentially is a straight line, as shown, over some range of values. Now raise the question: What prevents extrapolating that calibration? We believe it is nonlinearity. For the univariate case it is well-nigh self evident. At least it is to us — see Figure 1b. As Figure 1b shows, if the underlying data are linear, there should be no problem extending the calibration line (the extension being shown as a broken line), and using the extended line to perform the analysis with the same accuracy as the original data were analyzed. Yet, to not be able to extrapolate a calibration is something "everyone knows." What nobody knows, near as we can tell, is why we have to put up with that limitation.
There are a couple of other, low-probability answers that could be brought up, such as some sort of discontinuity in one or the other of the variables, but otherwise, any deviation of the data in the region of extrapolation would ipso facto indicate nonlinearity. Therefore, by far the most common cause of not being able to extrapolate that calibration is nonlinearity (almost by definition: a departure from the straight line is essentially the definition of nonlinearity). Engineers can point to various known physical phenomena of instruments to explain where nonlinearity in spectra can arise — stray light at the high-absorbance end and detector saturation effects at the low-absorbance end of the ranges, for example. Chemists can point to chemical interactions between the components as a source of nonlinearity at any part of the range. But mathematically, if you can make those effects go away, there is no reason left why you couldn't reliably extrapolate the calibration model.
Now let's consider a two-wavelength model for one of the components in a solution containing two components in a nonabsorbing solvent (hypothetical case, NOT water in the NIR!). The effect of nonlinearity in the relationship of the two components to their absorbances, will have different effects. If the component being calibrated for has a nonlinear relationship, that will show up in the plot of the predicted versus actual values, as a more or less obvious curvature in the plot, somewhat as shown in Figure 1b in our previous column (1).
A nonlinear relationship in the "other" component, however, will not show up that way. Let's try to draw a word picture to describe what we're trying to say here (the way we draw, this is by far the easier way): because we could imagine this being plotted in three dimensions, the nonlinear relation will be in the depth dimension, and will be projected on the plane of the predicted-versus-actual plot of the component being calibrated for. In this projection, the nonlinearity will show up as an "extra" error superimposed on the data, and will be in addition to whatever random error exists in the "known" values of the composition. Unless the concentrations of the "other" component are known, there is no way to separate the effects of the nonlinearity from the random error, however. While we can't actually draw this picture, graphical illustration of these effects have been published previously (8).
Again, however, if there is perfect linearity in the relationship of the absorbance at both wavelengths with respect to the concentrations of the components, you should be able to extrapolate the model beyond the range of either or both components in the calibration set equally well, just as in the univariate case.
The problem is knowing where, and how much nonlinearity exists in the data. Here is where Brown has made a good start on determining this property in his paper back in 1993, at least for the limited case he is dealing with: a designed experiment with (optically) nonscattering samples.
Now for Philip Brown's main (by our reckoning) result: when he used the wavelengths of minimum nonlinearity to perform the calibration at, he found that he was indeed able to extrapolate the calibration. Repeat: in circumstances under which the effects of data nonlinearity (from all sources) are minimized, he was able to extrapolate the calibration. In this paper he makes the statement, "One might argue that trying to predict values of composition outside the data used in calibration breaks the cardinal rule of not predicting outside the training data." He seems almost surprised at being able to do that.
But given our discussion earlier in the text, he should not be. So in this case, it is not only not surprising that he is able to extrapolate the predictions — we think that it is inevitable, because he has found a way to utilize only those wavelengths where nonlinearity is absent. Now what we need are ways to extend this approach to samples more nearly like "real" ones. And if we can come up with a way to determine the spectral regions where all components are linearly related to their absorbances, the issue of not being able to extrapolate a calibration should go away. Surely it is of scientific as well as practical and commercial interest to understand the reasons we cannot extrapolate calibration models. And then devise ways to circumvent those limitations.
Obviously, however, someone needs to do more research on that topic. We have contacted Philip Brown and asked him about this topic. Unfortunately, linearity per se is not of interest to him; the emphasis of the paper he wrote was on role of linearity in the wavelength-selection process, not the nonlinearity itself. Furthermore, in the ten years since that paper appeared, his interests have changed and he is no longer pursuing spectroscopic applications.
But to extend the work to understanding the role of nonlinearity in calibration, how to deal with it when an experimental design is not an option, and what to do when the optical scatter is the dominant phenomenon in the measurement of samples' spectra still are very open questions.
Jerome Workman Jr. serves on the Editorial Advisory Board of Spectroscopy and is director of research, technology, and applications development for the Molecular Spectroscopy & Microanalysis division of Thermo Electron Corp. He can be reached by e-mail at: jerry.workman@thermo.com Howard Mark serves on the Editorial Advisory Board of Spectroscopy and runs a consulting service, Mark Electronics (Suffern, NY). He can be reached via e-mail at: hlmark@prodigy.net.
References
1. H. Mark and J. Workman, Spectroscopy 20(1), 56–59 (2005).
2. H. Mark and J. Workman, Spectroscopy 20(3), 34–39 (2005).
3. H. Mark and J. Workman, Spectroscopy 20(4), 38–39 (2005).
4. H. Mark and J. Workman, Spectroscopy 20(9), 26–35 (2005).
5. H. Mark and J. Workman, Spectroscopy 20(12), 96–100 (2005).
6. H. Mark and J. Workman, Spectroscopy 2(9), 37–43 (1987).
7. H. Mark and J. Workman, Statistics in Spectroscopy (Academic Press, New York, NY, 1991).
8. H. Mark, Applied Spectroscopy 42(5), 832–844 (1988).
9. H. Mark and J. Workman, Spectroscopy 13(6), 19–21 (1998).
10. H. Mark and J. Workman, Spectroscopy 13(11), 18–21 (1998).
11. H. Mark and J. Workman, Spectroscopy 14(1), 16–17 (1998).
12. H. Mark and J. Workman, Spectroscopy 14(2), 16–27,80 (1999).
13. H. Mark and J. Workman, Spectroscopy 14(5), 12–14 (1999).
14. H. Mark and J. Workman, Spectroscopy 14(6), 12–14 (1999).
15. P. Brown, J. Chemom. 7, 255–265 (1993).
16. H. Mark and J. Workman, Spectroscopy 5(9), 47–50 (1990).
17. H. Mark and J. Workman, Spectroscopy 6(1), 13–16 (1991).
18. H. Mark and J. Workman, Spectroscopy 6(4), 52–56 (1991).
19. H. Mark and J. Workman, Spectroscopy 6 (July/August), 40–44 (1991).









LIBS Illuminates the Hidden Health Risks of Indoor Welding and Soldering
April 23rd 2025A new dual-spectroscopy approach reveals real-time pollution threats in indoor workspaces. Chinese researchers have pioneered the use of laser-induced breakdown spectroscopy (LIBS) and aerosol mass spectrometry to uncover and monitor harmful heavy metal and dust emissions from soldering and welding in real-time. These complementary tools offer a fast, accurate means to evaluate air quality threats in industrial and indoor environments—where people spend most of their time.

Smarter Sensors, Cleaner Earth Using AI and IoT for Pollution Monitoring
April 22nd 2025A global research team has detailed how smart sensors, artificial intelligence (AI), machine learning, and Internet of Things (IoT) technologies are transforming the detection and management of environmental pollutants. Their comprehensive review highlights how spectroscopy and sensor networks are now key tools in real-time pollution tracking.

New AI Strategy for Mycotoxin Detection in Cereal Grains
April 21st 2025Researchers from Jiangsu University and Zhejiang University of Water Resources and Electric Power have developed a transfer learning approach that significantly enhances the accuracy and adaptability of NIR spectroscopy models for detecting mycotoxins in cereals.




